

TRANSMITEREA INFORMAŢIEI EREDITARE
Informaţia ereditară, necesară pentru sinteza diferitelor proteine şi realizarea caracterelor fenotipice, se transmite din generaţie în generaţie, în două etape (vezi figura 1.1):
biosinteza unor noi molecule de ADN identice cu molecula iniţială – prin replicare semiconservativă – care va conduce la dublarea cantităţii de ADN;
distribuţia totală, egală şi precisă a materialului genetic dublat, prin diviziune celulară.
Aceste procese, riguros controlate, se realizează de obicei cu mare fidelitate (exactitate) asigurând astfel stabilitatea proceselor ereditare. Evident, ele pot suferi şi erori (de replicare sau de distribuţie) care, în absenţa unor mijloace eficace de reparare şi eliminare a erorilor, pot avea consecinţe importante la descendenţi, producând boli.
Transmiterea informaţiei şi caracterelor ereditare în succesiunea generaţiilor de celule şi organisme eucariote este supusă unor legi obiective şi universale.
REPLICAREA ADN.
Descriind modelul structurii ADN, alcătuit din două catene polinucleotidice, antiparalele (cu polaritate inversă), legate între ele (prin punţi de hidrogen) în mod complementar (A-T; G-C), Watson şi Crick au intuit de la început că modelul propus de ei explică mecanismul de copiere a informaţiei genetice: fiecare catenă serveşte drept matriţă pentru formarea unei catene noi.
Schematic, dubla spirală a ADN se separă în cele două catene componente (asemenea deschiderii unui fermoar), formând o structură în formă de Y numită “furcă de replicare”. Fiecare catenă serveşte apoi ca matriţă sau tipar pentru aranjarea complementară (A-T, G-C) şi secvenţială (în direcţia 5’→3’) a deoxiribonucleotidelor activate, care vor fi polimerizate sub acţiunea ADN polimerazei (figura 5.1.). Astfel, o moleculă bicatenară de ADN va forma două molecule noi, identice între ele precum şi cu molecula “parentală”, fiecare formată dintr-o catenă "veche" şi o catenă "nou sintetizată"; de aceea procesul de sinteză al ADN a fost numit replicare semiconservativă. În felul acesta informaţia genetică a fost transmisă întocmai, în succesiunea generaţiilor de molecule.
Experimental, s-a dovedit[1] că procesul real al replicării genomului este concordant cu această ipoteză dar prezintă un nivel de complexitate care a evoluat de la organismele inferioare la cele superioare. Cele mai multe studii ale mecanismului de replicare a ADN au fost efectuate la procariote, din cauza facilităţilor experimentale, dar mecanismul de bază este similar şi la eucariote. Cu toate acestea replicarea ADN în celulele animale este un proces foarte complex datorită mărimii enorme a genomului, fragmentării sale în cromosomi, asocierii cu proteine histonice în nucleosomi şi compactării, prin spiralizare, în fibre de cromatină. De aceea, replicarea ADN implică numeroase interacţiuni proteină-proteină şi proteine-ADN şi este precis reglată temporal şi spaţial, în concordanţă cu fazele ciclului celular. Pentru a putea discuta aceste “particularităţi” de complexitate, decisive pentru acurateţea transmiterii informaţiei ereditare, vom prezenta mai întâi “maşinăria” de sinteză şi mecanismul molecular “de bază” al replicării ADN.
1. PROTEINELE IMPLICATE ÎN REPLICAREA ADN
Replicarea ADN se face sub acţiunea precisă şi rapidă a unui complex multiproteic (numit şi sintesom), o veritabilă “maşină de replicare”, care se mişcă în lungul ADN (figura 5.2). Fiecare piesă componentă are funcţii precise.
ADN polimerazele. La om au fost identificate cinci tipuri de ADN polimeraze, denumite α, β, γ, δ şi ε; în replicarea ADN nuclear sunt implicate ADN polimeraza δ, enzima replicativă majoră, şi ADN polimeraza α; ADN polimeraza γ realizează replicarea ADN mitocondrial iar ADN polimerazele β şi ε participă la repararea ADN.
ADN polimerazele folosesc catena matriţă şi orientarea legăturilor de hidrogen ale purinelor şi pirimidinelor pentru a “stabili” care deoxiribonucleotid va fi adăugat în catena nouă. ADN polimerazele catalizează formarea legăturilor fosfodiesterice între deoxiribonucleotidele adiacente care vor forma catena de ADN. În legătură cu activitatea ADN polimerazelor sunt important de subliniat două caracteristici generale:
toate ADN polimerazele sintetizează ADN numai în direcţia 5’→3’, deci fiecare nou nucleotid va fi adăugat la gruparea 3’OH a nucleotidului deja încorporat într-o catenă polinucleotidică, de regulă catena în curs de sinteză;
ADN polimeraza “nu ştie” să înceapă sinteza unei catene ci numai să o alungească, adăugând un nucleotid la extremitatea 3’OH a unui acid nucleic (indiferent dacă este ADN sau ARN) care este legat complementar prin intermediul punţilor de hidrogen la catena matriţă; de aceea sinteza unei catene noi de ADN începe de fapt cu sinteza unei amorse de ARN (!) (în engleză primer).
ADN helicazele desfac dublu helixul de ADN (folosind energia furnizată de ATP) şi “eliberează” monocatenele ce vor funcţiona ca matriţe pentru replicare; ele acţionează în puncte specifice, precis localizate, numite şi origini ale replicării, şi apoi asigură înaintarea furcii de replicare, prin ruperea legăturilor de hidrogen, debobinarea şi deschiderea moleculei de ADN, care devine accesibilă proteinelor care iniţiază replicarea.
ADN topoisomerazele I şi II despiralizează helixul ADN şi eliberează tensiunea din molecula de ADN, acumulată prin debobinarea sa de către helicaze (care produc o superînrulare a ADN în aval de locul de acţiune). Topoisomerazele secţionează una sau ambele catene ale ADN (la nivelul axului fosfodiesteric), le derulează (relaxând molecula de ADN, prea strâns înfăşurată) şi apoi resudează breşa din fiecare catenă.
Există două tipuri de asemenea enzime: topoizomerazele I determină secţionarea unei singure catene ADN, iar topoizomeraza tip II determină rupturi ADN simultan la nivelul celor doua catene. In plus, topoizomeraza II pare a interveni şi în alte procese celulare precum condensarea cromosomilor în cursul mitozei şi separarea anafazică a cromatidelor surori.
Proteinele de replicare A (RPA) numite şi proteine SSB (de la single-strand binding proteins) menţin separate cele două catene desfăcute de helicaze, prin fixare la monocatene şi “mascarea” perechilor de baze, care au tendinţa naturală de a se reuni, prin refacerea punţilor de hidrogen; RPA împiedică reîmperecherea bazelor.
Complexul ADN primază – ADNpolimerază α este implicat în sinteza amorselor şi replicarea catenei “întârziate” a ADN. Sub acţiunea ADN primazei se sintetizează amorse scurte (3-10 nucleotide) de ARN la care ADN polimeraza α adaugă, la capătul 3’ terminal, circa 30 de deoxiribonucleotide. Apoi complexul ADN primază-ADN polimeraza α este îndepărtat şi ADN polimeraza δ “preia ştafeta” continuând sinteza lanţului de deoxiribonucleotide început. Amorsele ARN vor fi distruse prin hidroliză enzimatică şi înlocuite cu ADN.
Proteinele de replicare C (RPC) recunosc specific şi se leagă la joncţiunea dintre primer si matriţă, fiind responsabile de medierea şi stabilizarea interacţiunii ADN polimerazei cu matriţa ADN.
PCNA (de la Proliferating Cell Nuclear Antigen) se leagă în imediata vecinătate a proteinelor RPC şi, împreună, sunt principalele proteine asociate ADN polimerazei. PCNA se organizează sub forma unor dimeri care au configuraţia spaţială a unui inel ce lunecă de-a lungul matriţei ADN şi permit încorporarea neîntreruptă a mii de nucleotide în catena ADN nou sintetizată. Blocarea PCNA (de exemplu, prin proteina p21) opreşte replicarea ADN.
Ribonucleaza H1 (RNaza H1) îndepărtează amorsele ARN[2] folosite de ADN polimeraze pentru iniţierea replicării; lacuna rezultată este completată, refăcută, de ADN polimerază δ iar refacerea continuităţii catenei este realizată prin sudarea capetelor, de către o ADN ligază.
2. Mecanismul molecular al replicării ADN.
Procesul de replicare începe în “puncte” definite ale genomului numit origini de replicare (“ori”) şi de aici progresează în ambele direcţii, până ce ADN este complet duplicat. La nivelul originilor replicării se fixează pe ADN proteinele de iniţiere ale replicării – care formează compexele prereplicative (pre-RC) – de care se leagă apoi celelalte enzime ale “aparatului de replicare”: topoisomerazele, helicazele, primazele, ADN polimerazele.
La nivelul originilor replicarii, în cursul fazei G1 a ciclului celular, are loc formarea asa-numitelor complexe prereplicative (pre-RC) prin asamblarea secvenţială a unor proteine (ORC, Cdc6, Cdt1 si Mcm2-7). Odată format, complexul va fi activat prin intervenţia unor kinaze ciclin-dependente care declanşează tranziţia către replicarea ADN. Aceasta tranziţie implică asamblarea succesivă a unor factori de replicare adiţionali (precum Mcm10, CDK, DDK, Cdc45, Sld3 şi proteinele de replicare A) care facilitează despiralizarea ADN la nivelul originilor replicării şi culminează cu asocierea ADN polimerazelor la ADN-ul despiralizat.
Dubla elice este apoi despiralizată sub acţiunea topoisomerazelor iar catenele sunt separate temporar de către ADN helicaze, într-o regiune localizată, formând o structură în formă de Y – furca de replicare. Pentru a menţine catenele desfăcute, pe fiecare catenă “eliberată” se fixează proteinele RPA (sau SSB), care împiedică re-împerecherea bazelor complementare şi refacerea spontană a dublului-helix (figura 5.2.).
Pe cele două catene “matriţă” are loc, în prezenţa ADN polimerazei δ, aranjarea secvenţială şi complementară a deoxiribonucleotidelor activate în prealabil (deci sub formă de nucleosid trifostaţi), pentru a aduce energia necesară reacţiei de polimerizare.
Polimerizarea nucleotidelor se face diferit pe cele două catene matriţă, datorită a două elemente caracteristice, menţionate anterior:
cele două catene ale ADN sunt antiparalele;
ADN polimeraza nu poate iniţia sinteza de novo a unui lanţ polinucleotidic prin legarea a două mononucleotide; ea este capabila doar sa inducă adiţia unui nucleotit la capatul 3'OH al unui lanţ polinucleotidic preexistent; de aceea, declanşarea replicării ADN necesită utilizarea unor amorse de tip ARN (“primer”) – sintetizate sub acţiunea primazei; sinteza se face aşadar numai în direcţia 5' ® 3' iar ADN polimeraza “ştie” numai să alungească catena în curs de sinteză.
Aceste două caracteristici determină “o asimetrie” în procesul de replicare: pe catena matriţă 5’®3’ sinteza catenei noi este continuă şi rapidă (“leading strand” sau catenă avansată), pe măsura desfacerii celor două catene, iar pe catena 3’®5' sinteza ADN este discontinuă, sub forma unor secvenţe scurte ( 100-1000 nucleotide) de ADN numite şi”piese Okazaki”[3], fiind mult mai lentă (“lagging strand” sau catenă întârziată) (figura 5.3). Ulterior amorsa este distrusă prin hidroliză şi înlocuită cu secvenţe de tip ADN, sub acţiunea ADN polimerazei δ iar fragmentele sunt unite de către o ligază. Pentru catena avansată amorsa este necesară numai la începutul replicării; în schimb pentru catena întârziată sinteza fiecărui fragment Okazaki necesită o amorsă.
Particularităţile replicării ADN la eucariote.
Mecanismele de replicare a ADN la eucariote sunt în general asemănătoare celor de la procariote. Apar totuşi anumite particularităţi determinate de: mărimea enormă a genomului, asocierea ADN cu proteinele în nucleosomi şi fibre de cromatină şi momentul replicării în ciclul celular (faza S). ADN nuclear este localizat în mai mulţi cromosomi, fiecare cu o structură nucleoproteică complexă. Acest fapt implică iniţierea replicării în mai multe puncte de origine, sinteza concomitentă a proteinelor histonice necesare menţinerii organizării cromosomilor, reglarea precisă temporo-spaţială a replicării în unităţile de replicare sau repliconi. Sinteza ADN nuclear depinde de multiple proteine enzimatice sintetizate în citoplasmă precum şi de transmiterea stimulilor extracelulari care declanşează replicarea. Există o reglare a sintezei ADN dependentă de ciclul celular.
3.1. Originile replicării şi repliconii.
Datorită lungimii mari a genomului, asocierii cu proteine în nucleosomi şi organizării în fibre de cromatină, viteza de replicare a ADN la eucariote este redusă (circa 50 nucleotide pe secundă comparativ cu 500 nucleotide pe secundă la procariote, unde ADN este “gol”, neasociat cu proteine). ADN trebuie însă să fie replicat integral în circa 8 ore; pentru a echilibra această situaţie “conflictuală”, replicarea ADN la eucariote debuteză în mai multe puncte de origine, ce corespund unor mici unităţi de replicare numite repliconi. Replicarea progresează bidirecţional pentru fiecare replicon (figura 5.3) şi, după ce se termină, repliconii fuzionează treptat până ce întreagul cromosom este duplicat (figura 5.4). Fiecare cromosom uman are puncte de origine multiple, situate la 150-200 kb iar întregul genom posedă circa 30.000 de puncte de origine.
Punctele de origine a replicării sunt reprezentate de secvenţe specifice de ADN (de circa 3000 nucleotide) care semnalează startul replicării şi permit asamblarea "aparatului de replicare". Aceste secvenţe nucleotidice specifice, în care se începe replicarea, conţin două categorii de elemente: elemente de tip A, putenic conservate la toate eucariotele, reprezentate de secvenţele de replicare autonomă sau ARS (de la autonomously replicating sequence) bogate în repetiţii AT[4] şi elemente de tip B, mai puţin conservate.
APARATUL DE REPLICARE.
La eucariote există o “maşină” de replicare ce conţine toate elementele menţionate anterior (vezi A.1). De subliniat că ADN polimeraza a acţionează pe catena întârziată (“lagging”) şi ADN polimeraza d atât pe catena avansată (“leading”) cât şi pe cea întârziată. În reglarea activităţii ADN polimerazelor sunt implicate: PCNA sau antigenul nuclear de proliferare celulară şi factorul de replicare C (RF-C), ambele cu roluri importante în controlul pornirii sau opririi replicării.
REPLICAREA ADN ŞI NUCLEOSOMII.
ADN-UL eucariotelor este asociat cu proteine histonice formând nucleosomii, structuri compacte în care ADN se înfăşoară (de două ori) în jurul unui octamer de histone (vezi capitolul 2.D.2.3). Se pune firesc întrebarea: cum se comportă nucleosomii înainte şi după replicare? Se presupune că atunci când furca de replicare ajunge în dreptul nucleosomului, ADN se derulează şi miezul histonic se desface în două jumătăţi care rămân ataşate la una din cele două molecule de ADN formate prin replicare; ele vor reface ulterior nucleosomul original. Pe cealaltă moleculă de ADN se vor constitui însă noi nucleosomi. Acest model este ipotetic dar s-a demonstrat cu certitudine că o dată cu sinteza ADN are loc în nucleu o sinteză importantă de histone necesare constituirii de nucleosomi noi.
Replicarea ADN are loc în faza S a ciclului celular.
Celulele umane realizează procesul de replicare într-o perioadă particulară a ciclului lor de viaţă, faza S. Toate celulele ce se divid trec prin acest ciclu celular alcătuit din patru faze G1, S, G2 – ce corespund interfazei – şi M sau faza mitotică.
In celulele de mamifere faza S durează circa 8 ore (pentru un ciclu celular de 24 de ore); este un timp scurt pentru replicarea întregului genom uman diplod de peste 6 miliarde de pb. La această constrângere temporală se adaugă obligaţia ca replicarea ADN să fie foarte precisă deoarece orice eroare de plasare a nucleotidelor în catena nou sintetizată produce mutaţii. Celula are mijloacele necesare pentru a se asigura că întregul genom este integral şi corect replicat în cursul fazei S. La aceasta se adaugă mecanisme multiple de identificarea şi corecţie a erorilor
Reglarea replicării ADN – se realizează prin interacţiuni coordonate între diferite proteine: ciclinele, Cdk, inhibitoriii Cdk , precum şi anumiţi factori de transcripţie (care activează punctele de origine ale replicării)(vezi 5.B.1):
Ciclinele[5] D şi E facilitează trecerea G1/S iar ciclina A asigură progresia prin faza S;
Cdk4 este o protein kinază activatoare;
Factorii de transcripţie: RLF (inclus în complexul de pre-replicare) – activează punctele de origine a replicării şi împiedică re-replicarea în cursul aceluiaşi ciclu iar E2F stimulează expresia genelor necesare intrării în faza S;
Inhibitorii kinazelor dependente de cicline sau CKI (proteinele p21, p27 ş.a.) formează al doilea nivel de reglare al intrării şi progresii celulelor prin faza S; ei blochează aceste procese (inhibând complexele Cdk-cicline şi PCNA) fie atunci când apar leziuni în ADN fie în cursul diferenţierii terminale a unui ţesut (însoţită de pierderea capacităţii de replicare şi diviziune).
Unităţile de replicare (ce includ 20-80 repliconi) nu sunt activate concomitent şi la întâmplare ci asincron şi într-o anumită ordine, în funcţie de structura cromatinei în care ele se găsesc. Cromatina puternic condensată (heterocromatina) este replicată tardiv, la sfârşitul fazei S, în timp ce eucromatina, mai decondensată, se replică precoce, la începutul fazei S. Deci regiunile din genom în care cromatina este mai puţin condensată şi deci mai accesibilă maşinăriei de replicare sunt replicate mai precoce; acestea sunt de regulă benzile R (bogate în G-C) regiuni de ADN care conţin în special genele "domestice" (active în toate celulele). Regiunile din cromosomi care sunt replicate tardiv coincid cu benzile G, bogate în A-T.
In mod normal, în faza S întregul genom este replicat o singură dată. Deoarece replicarea ADN este asincronă există riscul teoretic ca unele regiuni să fie replicate repetat. Acest lucru nu se întâmplă deoarece o serie de factori inhibitori se fixează pe cromatina replicată şi nu permit replicarea ei repetată, până ce celula nu trece prin mitoză. Aceşti factori sunt îndepărtaţi prin diviziune şi celulele rezultate pot începe un nou ciclu, cu faza S, în care se produce o nouă replicare.
REPLICAREA TELOMERELOR
Telomerele sunt structuri cromatiniene specializate, situate la capetele cromosomilor eucariotelor, care asigură stabilitatea cromosomilor şi împiedică unirea cromosomilor prin capetele lor.
Telomerele cromosomilor umani, alcãtuite din ADN şi proteine, au o structurã identicã la specii diferite şi puternic conservatã în cursul evoluţiei (acest fapt atestã importanţa lor funcţionalã deosebitã). Telomerele sunt formate din elemente repetitive hexanucleotidice (la om TTAGGG) dispuse in tandem, care se extind pe o lungime variabila de 5-20 kb, in functie de tesut si de varsta individului. Datoritã particularitãţilor de replicare ale capetelor ADN liniar din cromosomi (ce implică folosirea unor amorse degradabile pentru iniţierea replicării), la fiecare capãt 5' al catenelor nou sintetizate de ADN rămâne o mică regiune (corespunzătoare ultimei amorse) care nu este replicată (figura 5.3.b). Extremitatea moleculelor de ADN ramâne cu o prelungire monocatenară 3', protuberantă şi instabilă. Aceasta se pliazã (spre înapoi) şi formeazã o structurã "în ac de pãr", care stabilizeazã ADN[6]; efectul stabilizant este favorizat şi prin asocierea, la acest nivel, a unor proteine speciale (TRF1 Şi TRF2).
Un fenomen caracteristic telomerelor este pierderea inexorabilã a unor secvenţe de ADN la fiecare replicare (diviziune celularã). Deoarece, la capãtul 5' al catenelor de ADN nou sintetizate replicarea este incompletã şi la fiecare ciclu celular se pierd 25-200 pb. Aceasta duce la scurtarea progresivã a telomerelor şi, dupã un numãr de replicãri, la oprirea replicãrii şi diviziunii (în Go, prin acţiunea proteinelor p53). Telomerele ar fi "cãlcâiul lui Achile al dublei spirale a ADN" (Olovnicov, 1993). Datoritã eroziunii permanente a telomerelor, celulele umane au numãr limitat de diviziuni atât in vivo[7] cât şi in vitro. Se poate spune cã lungimea telomerelor este un indicator al istoriei replicative a celulelor şi poate fi consideratã un fel de "ceas molecular" care aratã potenţialul replicativ restant al celulelor normale; când telomerele ating o lungime minimã criticã celula înceteazã sã se dividã şi "ceasul" se opreşte.
Pierderea secvenţelor telomerice poate fi compensatã prin adiţia de novo a secvenţelor TTAGGG la capătul 3’ al unei catene de ADN, de cãtre enzima telomerazã, care stabilizeazã telomerele şi previne scurtarea lor. Telomeraza este o ribo-nucleoproteinã ce conţine o scurtã secvenţã de ARN, complementarã repetiţiilor telomerice (la om aceastã secvenţã este: 3'-CCCAAUCCC-5') ce funcţionează ca matriţă internă (figura 5.5). Telomeraza este un tip neobişnuit de revers transcriptazã (ADN polimerazã ARN dependentã) care adaugã la capãtul 3'(bogat în G) noi secvenţe de nucleotide telomerice, fãrã sã necesite o matriţã exogenã. Sinteza celeilalte catene complementare a telomerului , bogată în C, implică o amorsă+ADN polimeraza α, ce foloseşte ca matriţă catena telomerică bogată în C, care a fost deja sintetizată de telomeraza.
Telomeraza este exprimatã în celulele germinale[8] tinere (din gonade), în blastocist şi cele mai multe ţesuturi embrio-fetale în sãptãmânile 16-20 ale dezvoltãrii; Activitatea telomerazei scade progresiv în viaţa fetalã şi dispare postnatal, în aproape toate celulele somatice normale; excepţie fac celulele stem din mãduva oaselor. Dispariţia activitãţii telomerazice în celulele somatice dupã naştere determinã scurtarea progresivã a telomerelor (dupã fiecare divizune), odatã cu vârsta. Când se atinge (la o vârstã înaintatã) o lungime criticã minimã diviziunea celularã se opreşte şi începe senescenţa. Pierderea telomerelor genereazã de asemenea instabilitatea genomicã şi creşterea frecvenţei rearanjamentelor cromosomice.
Telomeraza poate fi însã reactivatã în procesele inflamatorii dar mai ales în celulele canceroase. Acest lucru opreşte scurtarea telomerelor, creşte stabilitatea lor (prin adãugarea secvenţelor repetitive hexanucleotidice la capetele cromosomilor) şi astfel stimuleazã proliferarea celularã. Componenta catalitică a telomerazei, numită TERT, a fost localizată pe cromosomul 5p15.33 si a fost clonată în 1999. Componenta ribosomală numită TERC este codificată de o genă scurtă (451 nucleotide) care a fost localizată pe cromosomul 3q21-q28.
Recent, mutaţii ale genei TERC au fost identificate la pacienţi care prezintă o formă transmisă autosomal dominant de discheratoza congenitală. La aceşti pacienţi s-a documentat o lungime semnificativ mai scurtă a telomerelor. Mutaţii ale TERC au fost identificate de asemeni şi la unii pacienţi cu anemie aplastică idiopatică. Şi în aceste cazuri lungimea telomerelor a fost gasită a fi redusă.
În ultimii ani se acordã un interes doesebit studiului corelaţiei dintre scurtarea telomerelor, senescenţa celularã, expresia telomerazei şi cancerelor umane. A apãrut ceea ce se cheamã "ipoteza telomer – telomerazã a îmbãtrânirii şi cancerului" (Caseta 5.1).
Caseta 5.1
Telomerele si senescenţã.
"Ipoteza telomericã a senescenţei" (Harley, 1991) se bazeazã pe faptul cã în celulele somatice normale lungimea secvenţelor telomerice repetitive scade cu vârsta (de la 20.000 pb la embrion, la circa 6000 pb la vârstnic). Acest fenomen a fost demonstrat atât in vitro cât şi in vivo. In consecinţã, lungimea telomerelor poate fi consideratã un indicator ce mãsoarã capacitatea proliferativã restantã a celulei iar scurtarea progresivã a telomerelor (prin replicarea incompletã) – un biomarker al îmbãtrânirii. Astfel, în sindroamele de îmbãtrânire acceleratã – în sindromul Werner, sindromul Hutchinson-Gilford şi alte stãri progeroide – telomerele sunt mult mai mici decât la persoane normale de aceeaşi vârstã.
Nu credem cã relaţia dintre scurtarea telomerelor şi senescenţã este mecanismul fundamental (şi nici decum unic) al îmbãtrânirii. Dar ipoteza este plauzibilã, ca o componentã a îmbãtrânirii celulare, şi studiul ei poate aduce multe surprize şi, de ce nu, acţiuni benefice. De ex., (1) încetinirea ratei de scurtare a telomerelor în celulele şi ţesuturile normale ar putea afecta profund vârsta debutului şi severitatea multor boli asociate cu vârsta; (2) alungirea ex vivo a telomerelor în limfocitele T, fibroblastele dermice sau celulele endoteliale ar putea avea un impact major asupra rãspunsului imun în HIV, vindecarea rãnilor şi poate în bolile cardiovasculare
Telomerele, telomeraza şi boala canceroasã.
In mod cert telomerele asigurã protecţia şi stabilitatea cromosomilor. Pierderea telomerelor produce rearanjamente cromosomice sau poate duce la pierderea cromosomului respectiv din celulã. Aceasta ar putea explica pierderea heterozigozitãţii locilor pentru genele supresoare ale creşterii tumorale, un mecanism fundamental în cancerogenezã. In plus, s-a constatat că în cancer se produce o reactivarea a telomerazei, care stabilizeazã lungimea telomerelor, adãugând secvenţele hexamerice specifice, şi celulele încep sã se dividã activ. Acest fenomen este, foarte probabil, implicat în imortalizarea celularã[9] şi/sau creşterea continuã a tumorilor (favorizând poate şi metastazarea). Desigur relaţia dintre reactivarea telomerazei şi cancer nu este simplã. In primul rând existenţa realã a unor tumori telomerazo-negative aratã însã cã activarea telomerazei nu este obligatorie pentru formarea / creşterea tumorilor[10]; existã foarte probabil şi alte mecanisme de imortalizare celularã, independente de telomerazã. In al doilea rând, producerea cancerului necesitã şi alte mecanisme celulare, alte mutaţii adiţionale care sã determine invazia şi metastazarea. In al treilea rând nu se cunosc mecanismele moleculare de reactivare a telomerazei. Un exemplu este gena c-MYC care a fost identificata ca reglator esential al activitatii TERT. c-MYC interactioneaza cu promotorul genei TERT, iar mutatiile cu castig de functie ale genei c-MYC pot explica cel putin partial reactivarea activitatii telomerazice in unele cancere. Se presupune cã ar exista gene implicate în represia normalã a telomerazei postnatal, care prin mutaţie ar produce reactivarea enzimei. Nu se cunoaşte încã rolul telomerelor şi telomerazei în transformarea de la tumori benigne la tumori maligne.
Cu aceste precizãri şi pãstrând un entuziasm ponderat se poate afirma cã determinarea activitãţii telomerazei este un marker pentru diagnosticul precoce al cancerului şi pentru evaluarea prognosticului iar folosirea unor inhibitori ai telomerazei (în special molecule antisens[11] complementare faţă de componenta ARN a telomerazei umane) ar putea deveni un tratament eficace contra cancerelor. In concluzie, "telomeraza îndeplineşte multe din criteriile pentru o metodã idealã în terapia cancerului şi deşi existã încã multe întrebãri fãrã rãspuns despre ipoteza telomericã, optimismul pare justificat" (Shay et al., 1996).
Cert este cã ipoteza telomer – telomerazã a îmbãtrânirii şi cancerului, chiar dacã nu pare perfectã este seducãtoare şi în anii care vin vom mai auzi cu siguranţã multe lucruri noi, care sperãm sã fie cât mai benefice practicii medicale în gerontologie şi oncologie.
FIDELITATEA REPLICĂRII ADN.
Acurateţea replicării ADN este critică pentru transmiterea informaţiei ereditare în succesiunea celulelor şi organismelor. De aceea, organismul uman posedă numeroase mecanisme care determină ca rata erorilor de împerechere (“mismatch”) apărute în cursul procesului de replicare să fie extrem de redusă, de circa o încorporare greşită la 109 – 1010 nucleotide.
În momentul în care cele două catene ADN sunt separate, nucleotidele activate se aranjează "spontan" şi pe bază de complementaritate pe întreaga porţiune a matriţei de ADN astfel expusă. Stoichiometria reacţiei de aşezare pe bază de complementaritate permite împerecheri greşite (în special unele precum G-T) cu o probabilitate de 1 la 10.000. Gradul crescut de fidelitate al replicării este rezultatul unor activităţi specifice ale ADN polimerazelor.
Un prim mecanism prin care ADN polimerazele ajută la cresterea fidelităţii replicarii este prin inserţia corectă a bazelor în catena ADN nou sintetizată. ADN polimerazele nu catalizează încorporarea pasivă a oricărui nucleotid care este deja legat prin punţi de hidrogen la catena matriţă, ci discriminează activ împotriva unor împerecheri greşite probabil pe baza de conformaţie tridimensională. Recunoaşterea erorii se face pe seama distorsiunii pe care o produce în diametrul constant de 2nm al moleculei de ADN împerecherea greşită a două nucleotide cu purină (molecule mari) sau două nucleotide cu pirimidină (molecule mici). Această distorsiune acţionează ca semnal pentru îndepărtare noii baze inserate greşit. Mecansimul creşte acurateţea replicării de aproximativ 100 de ori, reducând rata aşteptată a erorilor de la 10-4 la aproximativ 10-6.
Un alt mecanism major responsabil pentru acurateţea replicării ADN este activitatea de autocorecţie (sau editare de la proofreading) a ADN polimerazelor. Aceasta este rezultatul capacităţii exonucleazice 3' – 5' care acţionează în sensul opus sintezei ADN şi înlătură nucleotidele încorporate greşit în catena nouă. Asemenea activitate exonucleazică 3' – 5' a fost documentată pentru ADN polimerazele delta si epsilon şi permite creşterea acurateţei replicării ADN de circa 100-1000 ori.
În sfârşit, puţinele erori de împerechere care apar totuşi în ciuda intervenţiei mecanismelor mai sus amintite vor fi ţinta unor mecanisme de reparare a ADN (în principal sistemul de reparare al erorilor de împerechere) care vor fi discutate în capitolul 6. Lipsa corectării lor va conduce la apariţia mutaţiilor, prin replicari celulare ulterioare. De exemplu, dacă în dreptul A de pe matriţă se va plasa eronat C, în loc de T, la următoarea replicare se va produce substituţia perechii de baze A-T cu C-G.
Cert este că integritatea structurală a ADN este extrem de importantă pentru supravieţuire şi toate celulele vii au mecanisme evoluate de reparare a leziunilor ADN. Această paradigmă a fost tragic confirmată prin descoperirea unor sindroame de predispoziţie la cancer care se produc prin mutaţii ale genelor ce codifică proteinele implicate în reparare. Astfel de mutaţii ale genelor (numite firesc gene mutator) determină o creştere de peste 100 de ori a ratei mutaţiilor şi produc o predispoziţie individuală la cancer, în special cancer de colon.
B. TRANSMITEREA INFORMAŢIEI GENETICE
DE LA O CELULĂ LA CELULELE „FIICE”
În celulele somatice, materialul genetic dublat în interfază, se distribuie în mod egal şi total celulelor fiice, prin procesul de diviziune mitotică. Rezultă două celule noi („fiice”), identice din punct de vedere genetic, atât între ele, cât şi cu celula („mamă”) din care provin. Procesele prin care o celulă îşi replică materialul genetic, îl divide egal şi îl transferă celulelor fiice se desfăşoară într-o ordine progresivă, precis reglată, ce formează ciclul celular. Controlul ciclului celular decide în ultimă instanţă dacă o celulă îşi va continua progresia prin ciclu (va creşte şi se va divide) sau va suferi o diferenţiere sau va trece într-o stare de repaus proliferativ. Pierderea controlului ciclului va genera o creştere celulară anormală (celule tumorale, defecte de dezvoltare) sau va duce la moarte celulară programată (apoptosis). Se pot produce de asemenea erori în segregarea cromosomilor în mitoză care vor genera anomalii cromosomice. Pentru aceste raţiuni, cunoaşterea proceselor ce alcătuiesc ciclul celular prezintă o importanţă deosebită pentru înţelegerea unor mecanisme patogenice generatoare de boli.
1. CICLUL CELULAR.
Ciclul celular reprezintă succesiunea de evenimente biochimice şi morfologice care se produc în viaţa unei celule, din momentul formării şi până la sfârşitul diviziunii sale. Ciclul celular are două mari perioade: interfaza şi diviziunea (figura 5.6).
Interfaza reprezintă perioada cuprinsă între două diviziuni succesive, în care se desfăşoară toate activităţile specifice unei celule. Evenimentul cel mai important al interfazei este sinteza de ADN (replicarea), prin care se dublează cantitatea de material genetic (4C). Ea se produce într-o perioadă limitată a interfazei, denumită faza S. Datorită acestui proces, interfaza poate fi subdivizată în trei etape succesive: faza G1 (presintetică), faza S (de sinteză) şi faza G2 (postsintetică sau premitotică).
Diviziunea celulară sau faza M ("mitotică") este alcătuită dintr-o serie de procese secvenţiale prin care materialul genetic (ADN), replicat în interfază, se distribuie egal şi total (segregare cromatidiană) formând doi nuclei distincţi, iar celula se împarte în două celule fiice (citokineză); acestea vor fi identice cu celula din care au provenit (vezi capitolul 1, figura 1.1.B). Prin replicarea ADN-ului şi diviziune se asigură transmiterea fidelă a informaţiei genetice în succesiunea generaţiilor celulare.
Durata ciclului celular poate varia mult între diferite ţesuturi, datorită variabilităţii fazei G1, celălalte faze fiind relativ constante ca durată. Pentru o durată medie de 24 ore, duratele aproximative ale fazelor sunt: G1 = 10 ore, S = 9 ore, G2 = 4 ore, M = 1 oră.
Tabelul 5.1. Caracteristicile principale ale fazelor ciclului celular mitotic[12]
|
PERIOADA |
FAZA ŞI DURATA (ore) |
EVENIMENTE |
CANTITATE ADN |
ASPECT LA MICROSCOPUL ELECTRONIC |
|
Interfază |
G1 (10 h) |
Sinteză intensă de ARN şi Proteine |
2C |
2n cromosomi monocromatidieni |
|
|
S (9h) |
Sinteză de ADN şi histone |
4C |
|
|
|
G2 (4h) |
Sinteza proteinelor fusului de diviziune Sinteza factorului de declanşareal mitozei |
4C |
2n cromosomi bicromatidieni despiralizaţi |
|
Diviziune |
M (1h) |
Profază Metafază Anafază |
4C 4C
|
Cromosomi bicromatidieni condensaţi (vizibili la microscopul optic) |
|
|
|
Telofază |
2C 2C |
Cromosomi monocromatidieni |
1.1. FAZELE CICLULUI CELULAR MITOTIC
a. Faza G1
Faza G1 (în engleză gap – interval, lacună, gol – deoarece nu i se cunoştea semnificaţia) reprezintă faza de start a ciclului celular, pentru celulele stimulate (de factori de creştere) să se dividă (tabelul 5.1). Fiecare cromosom (puternic despiralizat) este monocromatidian, fiind alcătuit dintr-o singură moleculă de ADN. Cantitatea de material genetic este 2C molecule de ADN sub forma a 2n (46) cromosomi despiralizaţi. În prima parte a fazei G1 (G1A), se produce o sinteză intensă de substanţe (ARN, proteine) necesare creşterii şi funcţionării celulei. Se acumulează ARN şi proteine până la o concentraţie prag, numită punct de restricţie "R", după care celulele trec în subfaza G1B, fiind condiţionate să intre în faza S şi să se dividă (figura 5.6).[13]
În anumite condiţii (lipsa factorilor de creştere, prezenţa unor inhibitori ai sintezei proteinelor etc.), celulele aflate în subfaza G1A pot trece într-o fază de activitate metabolică redusă, numită faza G0 sau G1Q (de la "quiescence" ‑ repaus, linişte), în care rămân viabile şi pot supravieţui timp îndelungat. Dacă aceste condiţii restrictive dispar, celulele G0 pot reveni în G1 şi apoi pot progresa spre faza S, deoarece îşi păstrează capacitatea de diviziune. Fazele G1 şi G0 sunt două stări fiziologice distincte ale celulei.
Unele celule aflate în subfaza G1A (sub influenţa unor inductori ai diferenţierii) părăsesc definitiv ciclul celular şi trec în faza G1D, ce corespunde celulelor diferenţiate; ele nu se mai divid şi mor după un anumit timp.
b. Faza S
Faza S (în engleză "synthesis"‑ sinteză) se caracterizează prin sinteza de ADN (realizată prin replicarea semiconservativă) şi sinteza de histone (vezi capitolul 5.A). În această fază cantitatea totală de ADN (2C) trebuie să fie replicată complet dar numai o singură dată; se produce astfel o dublare a cantităţii de material genetic (4C), condiţie obligatorie pentru desfăşurarea diviziunii celulare. Numărul de cromosomi rămâne 46, dar fiecare cromosom va fi bicromatidian, alcătuit din două cromatide identice ("surori"), deci va conţine două molecule de ADN.
Replicarea ADN în faza S este asincronă: unele segmente de ADN (bogate în perechi de baze G‑C) se replică precoce, la începutul fazei S, iar alte segmente (bogate în perechi de baze A‑T) se replică tardiv, la sfârşitul fazei S. Informaţii precise despre sinteza/replicarea ADN pot fi obţinute fie prin autoradiografie (cu ajutorul unui izotop radioactiv, de regulă timidină tritiată T3H), fie prin utilizarea bromodeoxiuridinei (BrdU), un analog al timinei. Ele se vor încorpora în molecula nou sintetizată, marcând-o radioactiv, în primul caz, sau modificându-i configuraţia, în al doilea caz.
Uneori, în faza S a ciclului celular mitotic se produce un "schimb egal de material genetic între cromatidele surori" (SCE – în engleză ”sister chromatide exchange”) (Caseta 5.2)
Caseta 5.2
Schimburile dintre cromatidele surori
În faza S a ciclului celular mitotic se poate produce un "schimb egal de material genetic între cromatidele surori" (SCE – ”sister chromatide exchange”). Frecvenţa SCE este în mod normal de 7‑10 per metafază; ea creşte însă în anumite boli (sindroamele de instabilitate cromosomică), precum şi după expunerea organismului la substanţe mutagene, clastogene (ce rup cromosomii) sau carcinogene. Astfel, SCE reprezintă o metodă de evaluare a acţiunii mutagene a diferiţilor compuşi chimici din mediul ambiant.
SCE care poate fi evidenţiat prin tehnici speciale. Celulele în cultură efectuează două cicluri de replicare în prezenţa unui analog al timinei, numit 5‑bromodeoxiuridină (BrdU). BrdU se încorporează, în locul timinei, în moleculele de ADN nou sintetizat, modificând proprietăţile de colorare ale cromatidelor. Folosind soluţia Giemsa sau colorantul fluorescent Hoechst 33258 se colorează cromatida în care numai o catenă a încorporat BrdU (figura 5.8.). În zonele în care se produce SCE se observă aspectul particular ("arlechin") al cromatidelor, cu zone colorate alternând cu zone necolorate.
c. Faza G2
Faza G2 se caracterizează prin sinteza unor proteine specifice şi a unor mici cantităţi de ADN (necesare în procesul de "corectare" a erorilor de replicare). Fiecare cromosom este bicromatidian (cantitatea de ADN este 4C), dar despiralizat. Spre sfârşitul fazei G2 se activează/ sintetizează un "factor de declanşare al mitozei" (MPF), ce determină condensarea filamentelor de cromatină în cromosomi şi formarea fusului de diviziune.
O demonstraţie elegantă a existenţei MPF este fenomenul de "condensare prematură a cromosomilor", realizat prin fuziunea unei celule în diviziune (metafază) cu o celulă în interfază (figura 5.7.); se produce rapid o condensare precoce a filamentelor de cromatină interfazică, care în mod obişnuit, sunt puternic despiralizate şi deci invizibile la microscopul optic, iar membrana nucleară se dezasamblează.
În lipsa factorului de condensare celulele se opresc în faza G2 şi pot abandona ciclul celular, formându-se celule tetraploide (4n cromosomi); unele dintre ele devin celule binucleate (de exemplu cardiomiocitele adulte)(figura 5.6).
d. Faza M
Faza M corespunde diviziunii mitotice şi are o durată de aproximativ o oră. Ea începe cu diviziunea nucleului (mitoză) şi se termină cu diviziunea citoplasmei (citokineză). Subliniem din nou că, în această etapă, materialul genetic dublat în interfază (4C molecule ADN în 46 cromosomi bicromatidieni) segregă, adică se distribuie în mod egal şi total celulelor "fiice" (2C molecule ADN în 46 cromosomi monocromatidieni), care vor fi astfel identice cu celula din care provin. Procesul de "distribuţie" a materialului genetic prin diviziune se desfăşoară de obicei cu mare precizie asigurând fidelitatea transmiterii informaţiei genetice în succesiunea generaţiilor de celule. El poate suferi însă şi erori, care vor genera anomalii cromosomice.
1.2. EVOLUŢIA CELULELOR REZULTATE PRIN DIVIZIUNE
Celulele rezultate după diviziune pot evolua în trei direcţii: proliferare, diferenţiere şi trecerea în stadiul de repaus.
a. Proliferarea.
Celulele parcurg un nou ciclu şi se divid repetat; aceste "celule ciclice" alcătuiesc compartimentul proliferativ al organismului şi se găsesc în ţesuturile embrionare, măduva hematogenă, stratul bazal al epidermului, etc.
b. Diferenţierea.
Celulele părăsesc definitiv ciclul celular şi se transformă în celule specializate, cu anumite structuri şi funcţii, care nu se mai divid şi mor după un timp determinat. De exemplu: neuronii, celulele musculare, granulocitele, hematiile mature, etc.
c. Stadiul de repaus.
Unele celule (de exemplu: celulele "stem" sau suşă; limfocitele ş.a.) părăsesc ciclul celular în faza G1 şi rămân în faza G0, având o activitate metabolică mai redusă, dar păstrându-şi capacitatea de diviziune. Aceste celule formează compartimentul neproliferativ. În condiţii speciale, ele reacţionează la anumiţi stimuli din mediu (factori de creştere, unii hormoni, substanţe mitogene etc.) şi pot reintra în ciclul de diviziune. Un exemplu edificator îl reprezintă activarea limfocitelor T din sângele periferic sub acţiunea fitohemaglutininei (PHA); ele se transformă în limfoblaste, celule tinere, care se divid intens. Acest fenomen este utilizat pentru studiul cromosomilor prin culturi de limfocite (vezi capitolul 2.D.3).
1.3 Controlul ciclului celular.
Progresia ordonată şi desfăşurarea normală a ciclului celular sunt realizate prin reacţii biochimice în care multiple kinaze ciclin-dependente (Cdk)(1-7) sunt activate, prin fixarea unor proteine numite cicline (A-H). După activare, fiecare complex proteic Cdk-ciclină fosforilează anumite proteine sepecifice, necesare pentru reacţiile care au loc într-o anumită fază a ciclului. Apoi, complexul Cdk-ciclină poate fi inactivat determinând trecerea la faza următoare a ciclului celular; inactivarea se poate face fie prin degradarea proteolitică a ciclinei (de către complexul proteasomilor, în urma ubiquitinării) fie prin intervenţia unor molecule inhibitoare CKI[14] (proteine KIP: p21, p27 sau proteine INK: p15, p16).
Interacţiunea dintre activarea şi inactivarea activităţilor CDK, în diferite momente cheie, asigură progresia normală şi reglarea ciclului celular. Fiecare fază a ciclului are un control specific (figura 5.9), realizat:
în G1 de complexul Cdk4-ciclina D,
la tranziţia G1/S de complexul Cdk 2-ciclină E,
în faza S de complexul Cdk 2-ciclină A,
iar în fazele G2 şi M de complexul Cdc2-ciclină B.
Tranziţia de la o fază la alta a ciclului celular este controlată prin mecanisme specifice, care acţionează în anumite puncte de control şi verifică dacă anumite procese sunt terminate înaintea începerii altora sau dacă nu există alterări ale componentelor ”maşinăriilor” de replicare şi segregare cromatidiană. În cazul identificării unor devieri de la normal sau defecte se blochează progresia celulei în ciclul celular (prin inactivarea Cdk) şi se induce ”repararea” sau, dacă aceasta nu este posibilă, apoptoza (moartea celulară programată). Prin aceste mecanisme se asigură creşterea normală şi se elimină defectele, care nu vor trece astfel la celulele fiice.
Pe parcursul ciclului celular se produc următoarele evenimente corelate cu reglarea ciclului:
În faza “de start” G1 celulele reacţionează la stimuli externi (mitogeni, factori de creştere) formându-se complexul Cdk4-ciclină D, care fosforilează şi deci activează diferite proteine ce funcţionează în această fază. Printre acestea, un rol important îl are proteina Rb[15], care (după fosforilare) eliberează un factor activator (E2F) al transcripţiei genelor ce funcţionează în faza S şi realizează replicarea ADN.
Intrarea în faza S este determinată de un semnal activator, complexul Cdk2-ciclină E. Formarea acestui complex este însă blocată de inhibitorul p27 al Cdk; trecerea în faza S implică deci mai întâi degradarea proteolitică a p27 (prin sistemul ubiquitină-proteasomi)
În punctul de control G1/S are loc o verificare a parametrilor de evoluţie normală a celulei: orice alterare a ADN, depleţie de oxigen / metaboliţi / energie sau pertrurbare fiziologică declanşează sinteza proteinei tp53 (“gardianul” genomului uman)[16] care activează transcripţia genei ce codifică proteina p21, un inhibitor al complexelor Cdk-ciclină precum şi al PCNA (antigenul nuclear al celulelor proliferante)[17]. Celulele sunt oprite în G1, oferindu-li-se timp de corecţie (figura 5.10); dacă alterările (în special cele din structura ADN) nu sunt reparate, celula va fi direcţionată spre apoptoză
Progresia ulterioară prin faza S şi replicarea ADN sunt reglate de către CDK2-ciclina A[18].
În faza G2, la punctul de control G2/M, se decide dacă celula intră în mitoză; acest lucru este determinat de activarea bruscă a complexului Cdc2[19]-ciclină B (numit anterior şi factorul MPF, de la “Mitosis Promoting Factor”). Celulele cu aberaţii cromosomice sau defecte ale aparatului mitotic sunt oprite să intre în mitoză (prin acţiunea tp53 şi p21, care blochează formarea complexului Cdc2-ciclina B).
În cursul mitozei, în metafază, mai există un punct de control M în care diviziunea se opreşte şi este verificată alinierea perfectă a cromosomilor, înaintea separării cromatidelor surori. Acţiunea este realizată de către o proteină inhibitoare ISS; degradarea proteolitică a acestei proteine (determinată de ubiquitină) declanşează anafaza, prin separarea cromatidelor.
La sfârşitul mitozei are loc degradarea bruscă a ciclinei B (produsă de către ubiquitină) şi inactivarea Cdc2, fenomene ce permit celulei să treacă într-o nouă fază G1.
Proliferarea celulară este reglată pe o durată limitată de factori extracelulari (hormoni, factori de creştere etc.) precum şi de anumite gene de proliferare sau "mitogene" (ce codifică cicline, receptori ai factorilor de creştere, proteine necesare sintezei de ADN, etc.). Deoarece prin mutaţia şi activarea lor anormală se produce o proliferare celulară aberantă şi cancer, aceste gene normale mai sunt numite şi proto-oncogene. O altă categorie de gene, numite antiproliferative, au rolul de a inhiba proliferarea celulară. De aceea aceste gene mai sunt numite gene supresoare de tumori sau antioncogene.
În cancer se produce o perturbare a desfăşurării normale a ciclului celular. Celulele canceroase, purtătoare de mutaţii, "scapă" de sub controlul mecanismelor care reglează proliferarea normală, parcurg rapid şi repetat ciclul celular, multiplicându-se permanent şi anarhic. Multe medicamente anticanceroase (citostatice) determină "blocarea" proliferării celulelor, prin oprirea evoluţiei lor în diferite faze ale ciclului celular.
2. MITOZA
Mitoza este o diviziune caracteristică celulelor somatice ale organismului. Ea asigură creşterea biomasei organismului, care, între stadiul de zigot unicelular şi cel de organism adult necesită 1×1014 diviziuni celulare. În unele ţesuturi, cum ar fi măduva hematogenă sau ţesutul epitelial, mitoza se desfăşoară continuu, pe tot parcursul vieţii, asigurând reînnoirea celulară dar şi repararea unor leziuni tisulare. Prin mitoză, materialul genetic dublat în interfază, se distribuie total şi egal celulelor fiice. Mitoza este o diviziune ecuaţională, deoarece din celula iniţială (celula “mamă”) cu 46 cromosomi rezultă două celule “fiice” care au, de asemenea, 46 cromosomi (celule diploide). Mitoza permite transmiterea cu mare fidelitate a informaţiei genetice în succesiunea generaţiilor de celule, ceea ce asigură stabilitatea proceselor ereditare.
2.1. FAZELE MITOZEI
Mitoza este un proces continuu, care durează circa o oră, şi se desfăşoară în cinci etape succesive: profaza, prometafaza, metafaza, anafaza şi telofaza (figura 5.11). Fiecare din aceste faze sunt definite prin acţiunea cromosomilor şi distribuţia conţinutului citoplasmatic iar desfăşurarea lor este riguros controlată pentru a se asigura că fiecare celulă fiică va primi o copie exactă a genomului parental. Orice eroare de distribuţie va genera anomalii cromosomice la celulele fiice.
a. Profaza
La începutul profazei, în celule pot fi observate o serie de fenomene citoplasmatice şi nucleare.
În citoplasmă, centrosomul (un organit citoplasmatic perinuclear) se divide, iar cei doi centrosomi rezultaţi se deplasează în direcţii opuse, formând polii fusului de diviziune. Din fiecare centrosom se ansamblează radiar microtubulii, care formează fibrele fusului de diviziune, ce pot fi centriolo-cromosomiale şi centriolo-centriolare (figura 5.12.a). De asemenea, componentele microtubulare ale citoscheletului celular suferă transformări esenţiale, prin depolimerizare în dimeri de α-tubulină şi β-tubulină şi reansamblare în fibre ale fusului de diviziune. Alte componente, cum ar fi moleculele de γ-tubulină şi o proteină minoră – pericentrina formează prin ansamblare centriolii. Centriolii au un ciclu propriu de diviziune, după fiecare mitoză fiecare celulă fiică moşteneşte doi centrioli, iar în interfaza ce urmează ei se divid, astfel încât înainte de diviziune vor fi patru centrioli, grupaţi câte doi şi dispuşi perpendicular unul pe altul.
In nucleu are loc condensarea fibrelor de cromatină care se scurtează şi se îngroaşă, devin intens colorate şi vizibile la microscopul optic sub formă de cromosomi. Nucleolii diminuă ca mărime şi în cele din urmă se dezintegrează.
Fiecare cromosom este alcătuit din două cromatide surori, deoarece materialul genetic s-a replicat (dublat) în faza S a interfazei. Ele sunt ataşate la nivelul centromerului, o structură cromosomică esenţială pentru a asigura distribuţia (segregarea) fiecărei cromatide în celulele fiice.
Regiunile centromerice sunt bogate în secvenţe de ADN satelit α şi conţin cantităţi considerabile de heterocromatină constitutivă. Componenta centromerică cu rol în segregare este kinetocorul, care este situat în interiorul centromerului, având la examinarea prin microscopie electronică aspectul unui corpuscul fibros mai dens cu diametrul de 400 nm. Kinetocorul este structura de care se ataşază câte o fibră a fusului de diviziune. Locul unde se formează kinetocorul este dictat de o secvenţă specifică de ADN de circa 120 pb, dispusă într-o zonă rezistentă la acţiunea nucleazelor. Aceste secvenţe sunt similare la toţi cromosomii, deoarece experimental s-a constatat că prin schimbarea lor reciprocă între neomologi, funcţia de segregare se păstrează. Secvenţa de circa 120 pb din zona kinetocorului cuprinde trei tipuri de motive: a) CDE-I, formată din 9 pb, dispusă la capătul 5’ al secvenţei kinetocorice şi conservată cu mici variaţii; b) CDE-II, formată din 80-90 pb ce conţin în proporţie de peste 90% AT (la eucariotele superioare prezintă secvenţe scurte repetitive, ce permit câteva distorsiuni considerabile ale dublului helix), dispusă în mijlocul secvenţei kinetocorice; c) CDE-III, formată din 11 pb, dispusă la capătul 3’ şi înalt consevată. Importanţa acestor secvenţe este sugerată de consecinţele mutaţiilor la acest nivel. Astfel, mutaţiile în CDE-I şi CDE-II, duc la pierderea parţială a funcţiei centromerului, în timp ce mutaţiile punctiforme din porţiunea centrală a CDE-III duc la pierderea totală a funcţiei centromerului. De asemenea, pentru funcţia centromerului este important un complex proteic de 240 kD, format din trei polipeptide, denumit Cbf-III, cu rol de legare a secvenţei CDE-III. Mutaţiile în una din cele trei gene care codifică polipeptidele Cbf-III blochează segregarea cromatidelor surori.
Cromatidele conţin în regiunea centromerului o secvenţă specifică de ADN repetitiv, unde se asamblează complexe proteice specializate, numite kinetocori (figura 5.12.b). Fiecare cromatidă are un kinetocor, la care se va fixa un microtubul, ce leagă centromerul fiecărui cromosom de polii fusului de diviziune. Ataşarea cromatidelor este este rezultatul interventiei unor complexe proteice numite coezina si condensina.
b. Prometafaza
Prometafaza începe cu dezansamblarea membranei nucleare, datorată disocierii laminei nucleare în subunităţi de laminină. Simultan cu dezasamblarea membranei nucleare, are loc şi dezintegrarea reticulului endoplasmatic şi a aparatului Golgi în mici vezicole membranare, prin mecanisme insuficient clarificate. Dezintegrarea membranei nucleare este urmată de deplasarea fusului mitotic în arie pe care a ocupat-o nucleul. Unii microtubuli se ataşează la kinetocorii fiecărui cromosom, legând centromerul fiecărui cromosom de polii fusului de diviziune, ceea ce permite deplasarea cromosomilor spre ecuatorul celulei.
c. Metafaza
În metafaza propriu-zisă, cromosomii bicromatidieni se aliniază independent unul de altul la ecuatorul fusului de diviziune, în acelaşi plan, formând aşa-numita placă metafazică[20].
d. Anafaza
Anafază începe brusc cu separarea cromatidelor surori, simultan la toţi cromosomii, şi mişcarea lor spre polii opuşi ai fusului de diviziune. Prin acest proces de disjuncţie (segregare) cromatidiană, fiecare cromosom bicromatidian formează doi cromosomi monocromatidieni. După separare, cromosomii monocromatidieni sunt, în acelaşi timp, traşi spre polii fusului de diviziune prin scurtarea depolimerizarea) fibrelor centriolo-cromosomiale, respectiv prin alungirea (polimerizarea) fibrelor centriolo-centriolare.[21] Deplasarea cromosomilor spre poli se face simultan, cu aceeaşi viteză. Astfel, se produce împărţirea totală şi egală a materialului genetic între celulele fiice.
e. Telofaza
Se caracterizează prin evenimente opuse celor din profază: cromosomii suferă un proces de decondensare şi despiralizare, fusul de diviziune se dezasamblează, se refac membranele ce delimitează nucleul, reticulul endoplasmic şi aparatul Golgi. Începe diviziunea citoplasmei (citokineză) prin acţiunea continuuă a unui inel contractil de actină, care produce diviziunea citoplasmei, cele două celule fiice se separă, fiecare conţinând 2n cromosomi monocromatidieni cu aspect interfazic şi o cantitate aproximativ egală din masa citoplasmatică.
2.2. reglarea MITOZEI
Desfăşurarea mitozei este riguros controlată (prin 3 puncte de control) de către celula „mamă” pentru a se asigura că fiecare celulă „fiică” va primi o copie exactă a genomului parental şi o cantitate aproximativ egală din masa citoplasmatică. Orice eroare de distribuţie va genera anomalii cromosomice la celulele fiice.
La tranziţia G2/M există un punct de control (1) crucial, în care se decide dacă celula intră în mitoză, asupra căruia acţionează Cdc2 şi ciclina B. Activarea bruscă a complexului Cdc2-ciclină B (numit şi MPF de la M-phase promoting factor) determină iniţierea mitozei.
MPF acţionează prin activarea unor protein-kinaze sau prin fosforilarea directă a unor proteine responsabile pentru modificarile asociate mitozei. Printre proteinele ţintă se numara condensinele care sunt responsabile de declanşarea condensării cromosomilor, laminele a căror fosforilare determină fragmentarea membranei nucleare, proteinele GM130 a căror fosforilare induce degradarea aparatului Golgi şi a reticulului endoplasmatic şi proteinele asociate microtubulilor care induc formarea fususlui de diviziune.
Dacă există o alterare cromosomică sau un defect al aparatului mitotic este activată proteina TP53, care induce sinteza proteinei 21, un inhibitor al Cdk, ce blochează celula în G2.
După ce fiecare cromosom s-a aliniat în placa metafazică procesul se opreşte (circa 20-30 minute) într-un nou punct de control (2), pentru a se verifica dacă toţi cromosomii sunt perfect aliniaţi înaintea separării cromatidelor surori (figura 5.13). Dacă totul este „perfect” atunci se declanşează degradarea proteolitică a proteinelor (coezina si condensina) ce ataşează cele două cromatide la nivelul centromerului, sub acţtiunea unui complex proteic cu activitate enzimatică numit separază care a fost activat de către complexul de promovare al anafazei, prin îndepartarea aşa-numitei securine, o componentă inactivatoare. Aceste fenomene permit separarea cromatidelor surori şi iniţierea anafazei.
În anafază există punctul de control (3) al mitozei când se produce inactivarea complexului Cdc2-ciclină B, prin degradarea proteolitică bruscă a ciclinei B (de către ubiquitină) şi inactivarea Cdc2, fapt ce va permite celulei terminarea mitozei şi trecerea într-o nouă fază G1.
2.3. ERORI DE DISTRIBUŢIE A MATERIALULUI GENETIC În MITOZĂ
Principalele erori de distribuţie a materialului genetic în mitoză sunt: nedisjuncţia cromatidiană, întârzierea anafazică şi clivarea transversală a centromerului. Ele generează anomalii cromosomice în ţesuturile somatice (vezi capitolele 6.B.1 şi 10.C)
a. Nedisjuncţia cromatidiană
Nedisjuncţia cromatidiană apare atunci când cele două cromatide ale unui cromosom nu se separă în timpul anafazei mitozei, ci rămân unite şi migrează împreună în una din cele două celule fiice. Consecinţa acestei erori este apariţia unor celule anormale: una cu un cromosom în plus (2n+1) = trisomică (47 cromosomi) alta cu cromosomul respectiv lipsă (2n‑1) = monosomică (45 cromosomi).
b. Întârzierea anafazică
Întârzierea anafazică constă în migrarea cu întârziere a unui cromosom monocromatidian, care în momentul formării membranelor nucleare va rămâne în afara nucleilor celulelor fiice. El formează un “micronucleu” care dispare la următoarea diviziune. Rezultă clone celulare cu 2n‑1/2n, adică 45/46 cromosomi. Prezenţa micronucleilor (ce se formează din cromosomi care nu au migrat şi care nu au fost incluşi în nici unul din nucleii fii), indică tulburări ale desfăşurării mitozei, ce pot fi produse de cauze externe. De aceea testul micronucleilor este utilizat pentru studiul efectelor cromosomice produse de radiaţiile ionizante sau alţi factori mutageni.
c. Clivarea transversală a centromerului
Clivarea transversală a centromerului produce isocromosomi (“cromosomi cu braţe egale”) alcătuiţi numai din braţe scurte (p) sau numai din braţe lungi (q)(figura 5.14). Aceştia sunt anormali, deoarece au duplicate genele de pe braţul prezent şi le lipsesc genele de pe braţul pierdut. În patologia umană au fost descrişi mai frecvent isocromosomi Xp sau Xq în disgeneziile gonadale sau i(12p) în sindromul Pallister-Killian, cu anomalii congenitale multiple şi retard mintal.
d. Absenţa citokinezei
Celulele somatice după duplicarea ADN-ului în interfază au 2n cromosomi bicromatidieni (4C). Dacă are loc mitoza dar nu se produce citokineza rezultă o celulă tetraploidă cu 4n cromosomi monocromatidieni (4C). Acest proces, numit şi endomitoză, se observă, în mod fiziologic, în procesul de regenerare hepatică.
e. Consecinţele erorilor mitotice
Erorile de distribuţie a materialului genetic în mitoză determină apariţia unor anomalii de număr şi structură ale cromosomilor în celulele fiice. Ele vor avea consecinţe asupra evoluţiei celulelor, influenţând viabilitatea şi capacitatea de multiplicare. Aceşti parametri depind mai ales de cromosomul afectat şi tipul de anomalie cromosomică (trisomie sau monosomie) produsă. Dacă celulele rezultate sunt viabile şi se multiplică ulterior, rezultă o clonă (grup de celule ce au o particularitate comună şi provin prin mitoze repetate dintr-o celulă iniţială modificată) iar în organism apar mozaicuri cromosomice (figura 5.15). Se întâlnesc mai frecvent în sindromul Turner (25% cazuri), sindromul Down (2-4% bolnavi) şi sindromul Klinefelter (vezi capitolul 10.C) Apariţia unor clone celulare anormale la un organism, prin oricare din erorile amintite, determină efecte diferite în raport cu momentul ontogenetic în care s‑a produs accidentul (şi deci cu proporţia celulelor anormale care se formează). Perturbarea precoce şi apariţia unui număr mare de celule anormale în primele stadii de dezvoltare are consecinţe negative majore (anomalii congenitale) asupra fenotipului purtătorilor de anomalii cromosomice (vezi capitolul 10.C). Un alt factor ce influenţează efectele fenotipice ale clonelor anormale este distribuţia lor în diferite ţesuturi. A fost descris, de exemplu, un mozaicism limitat la placentă, cu un făt euploid (vezi capitolul 18.3).
C. TRANSMIT EREA INFORMAŢIEI GENETICE
DE LA PĂRINŢI LA DESCENDENŢI
Transmiterea informaţiei genetice în succesiunea generaţiilor de organisme este un proces complex care se desfăşoară în două etape: formarea gameţilor, celule sexuale mature cu un număr haploid de cromosomi (23 la om) şi fecundarea lor, care reface la zigot numărul diploid de 46 de cromosomi, caracteristic speciei umane şi realizează o structură genetică nouă, unică şi constantă (individualitatea genetică) caracteristică viitorului individ. Ambele etape se efectuează de obicei cu mare precizie dar uneori se pot produce erori care generează anomalii genetice.
GAMETOGENEZA
Gametogeneza are loc în gonade, prin meioză – un tip special de diviziune reducţională – care asigură transmiterea informaţiei genetice la generaţia următoare şi îndeplineşte două funcţii majore:
Reduce la jumătate (n) numărul de cromosomi şi asigură astfel, după fecundare, menţinerea constantă a numărului de cromosomi ai speciei;
Creează diversitatea genetică ce apare la copii născuţi din aceiaşi părinţi[22], prin fenomenele de recombinare genică (din profaza I) şi recombinare intercromosomică (din anafaza I); meioza este principala sursă de variabilitate genetică.
CARACTERELE GENERALE ALE MEIOZEI
Meioza este un proces complex care se realizează prin două diviziuni succesive: meioza I (diviziunea meiotică primară, heterotipică, reducţională) şi meioza II (diviziunea meiotică secundară, homotipică, ecuaţională), neseparate de interfază, deci ADN se replică numai o singură dată.
a. Meioza I
Este o formă specială de diviziune celulară[23], unică şi foarte complexă, cu o durată lungă, în special la organismele feminine. Prezintă patru etape: profaza I, metafaza I, anafaza I, telofaza I, fiecare cu anumite particularităţi (figura 5.16.A).
(1). Profaza I este foarte lungă (90 % din durata meiozei I) şi cuprinde cinci stadii.
Leptoten. Prin condensarea fibrelor de cromatină, cromosomii devin vizibili ca filamente subţiri şi lungi, ataşate prin telomere la membrana nucleară. Astfel, datorită lungimii mari şi grosimii mici a cromosomilor, deşi cromosomii sunt bicromatidieni, ei apar monocromatidieni la examinarea la microscopul optic.
Zigoten. Cromosomii omologi (matern şi patern) se apropie, se dispun paralel de-a lungul cromatidelor, fenomen numit sinapsă sau conjugare cromosomică; se realizează astfel o aliniere "genă la genă" sau, mai exact, o împerechere între secvenţele omologe[24], rezultând structuri numite bivalenţi (în realitate fiecare unitate este o tetradă, deoarece prezintă patru cromatide). Cromosomii omologi sunt uniţi (lipiţi) în anumite regiuni, la nivelul complexului sinaptonemal, vizibil la microscopul electronic. O excepţie de la acest model de sinapsă, este prezentă la sexul masculin, la care cromosomii X şi Y, nefiind omologi, fac sinapsă "cap la cap", printr-o mică regiune omologă ("regiune pseudoautosomală") aflată la capătul braţelor scurte, şi formează prin condensare o structură specifică ‑ vezicula sexuală.
Pahiten. Cromosomii se scurtează şi se îngroaşă. Din loc în loc devin vizibile nişte puncte mai intens colorate numite cromomere, al căror număr şi poziţie sunt caracteristice fiecărui cromosom[25]. În faza de pahiten are loc fenomenul de încrucişare cromosomică între cromosomii omologi – crossing‑over – care constă în "ruperea" cromatidelor nesurori exact în acelaşi punct şi schimbarea reciprocă (încrucişată) a unor fragmente egale („recombinare omologă”)[26]; astfel, un fragment de cromatidă de pe un cromosom matern se transferă pe omologul său patern şi invers, realizând o nouă combinaţie genică cu material genetic de la ambii părinţi (vezi figura 3.4.). Fenomenul implică numai două din cele patru cromatide, care vor conţine gene de la ambii părinţi; se realizează astfel o recombinare genică (intracromosomică), care reprezintă o sursă de variabilitate genetică, datorită noilor combinaţii de gene care apar şi se transmit la descendenţi (vezi capitolul 3.A.2.2). La om se produc 1-3 recombinări per cromosom, în funcţie de dimensiunea cromosomului. În meioza masculină totalul recombinărilor este de circa 50 per celulă, în timp ce meioza feminină se caracterizează printr-un număr mai mare de recombinări (aproximativ 70 per celulă). Frecvenţa recombinării care se produce între două gene situate pe acelaşi cromosom depinde de distanţa dintre ele; calculul frecvenţei de recombinare permite alcătuirea hărţilor cromosomice, care indică localizarea genelor în lungul cromosomilor (vezi capitolul 3.D.1).
Diploten. Cromosomii omologi încep să se separe longitudinal, ca şi cum "s‑ar respinge" reciproc. Cromatidele lor rămân în contact la nivelul anumitor puncte ("chiasmata", pluralul de la "chiasma") care marchează localizarea crossing-over-ului.
Diakineza. Cromosomii devin mai scurţi şi mai groşi, omologii se separă aproape complet, iar chiasmatele se observă numai la capetele lor (terminalizarea chiasmatelor). În această fază se observă clar că fiecare bivalent conţine patru elemente: cromatidele surori sunt unite prin centromer, iar cromatidele nesurori sunt unite prin chiasmatele la nivelul cărora s‑a produs crossing‑over‑ul.
(2) Metafaza I. Membrana nucleară dispare complet şi se formează fusul de diviziune. Bivalenţii, formaţi din cromosomi bicromatidieni, se fixează cu centromerul pe filamentele fusului şi se deplasează spre placa metafazică, la ecuatorul celulei. Centromerul cromosomilor omologi se dispune aleatoriu de o parte şi de alta a planului ecuatorial. Metafaza I este etapa optimă de studiu a cromosomilor în meioză.
(3). Anafaza I cuprinde un fenomen genetic foarte important: disjuncţia cromosomică. Cei doi cromosomi bicromatidieni ai fiecărui bivalent se separă şi se repartizează câte unul la fiecare celulă fiică. Spre deosebire de mitoză, cromatidele surori nu se despart, ci rămân ataşate la nivelul centromerului. Apoi are loc migrarea anafazică prin deplasarea cromosomilor (bicromatidieni) simultan şi cu aceeaşi viteză, spre polii opuşi ai celulei. În final, se produce o reducere a numărului de cromosomi, de la 2n la n şi fiecare celulă va avea numai un exemplar din perechea de omologi. Acest fenomen stă la baza primei legi a lui Mendel, legea segregării.
Segregarea sau separarea aleatorie a fiecărei perechi de cromosomi omologi determină asortarea independentă a omologilor, fenomen denumit recombinare intercromosomică. Deoarece fiecare pereche de cromosomi se separă independent de celelalte perechi rezultă un număr mare de combinaţii cromosomice în gameţi, în raport direct cu numărul de perechi de cromosomi ai speciei (figura 5.17). Se formează 223 tipuri de gameţi, deci peste 8,4 milioane de gameţi diferiţi, la fiecare din cele două sexe.
Asortarea independentă a cromosomilor omologi, prin fenomenul de recombinare cromosomică, asociată cu recombinarea intracromosomică prin crossing-over, explică marea variabilitate a gameţilor. Ea confirmă cea de a doua lege a lui Mendel, referitoare la combinarea liberă a "factorilor ereditari" (genelor) astfel că fiecare gamet, care rezultă în urma meiozei, are un set unic de gene, diferit de al celorlalţi. Prin combinarea, pe bază de probabilitate, a gameţilor masculini cu cei feminini în procesul de fecundare are loc formarea unor zigoţi diferiţi din punct de vedere genetic, iar indivizii rezultaţi vor fi unicate genetice.
(4). Telofaza I. Are loc reasamblarea nucleilor; citokineza se realizează fără separarea completă a celulelor fiice, care rămân ataşate printr‑o punte citoplasmatică, formând o diadă.
b. Meioza II
Meioza II începe imediat după terminarea meiozei I şi se aseamănă cu o diviziune mitotică dar care se realizează în celule cu număr haploid de 23 de cromosomi bicromatidieni (dintre care unii sunt recombinanţi) şi nu este precedată de o replicare a ADN. Este deci o diviziune homotipică, ecuaţională. Ea cuprinde, de asemenea, cele patru stadii: profaza II, metafaza II, anafaza II şi telofaza II (figura 5.16. B).
(1). Profaza II: cromosomii bicromatidieni se individualizează, cromatidele surori devin vizibile distinct, iar membrana nucleară dispare.
(2). Metafaza II. Cromosomii se condensează şi se ataşează fiecare pe câte un filament al fusului de diviziune, aliniindu-se în placa metafazică ecuatorială.
(3). Anafaza II. Prin diviziunea centromerelor – disjuncţie cromatidiană – fiecare cromosom se separă în două cromatide; acestea devin cromosomi independenţi şi se deplasează spre polii opuşi ai celulelor.
(4). Telofaza II. Din cele două celule fiice se formează patru seturi haploide de cromosomi monocromatidieni (câte două pentru fiecare celulă). În final, prin reorganizarea nucleilor şi separarea celulelor rezultate apar patru celule haploide, care prin maturare se transformă în gameţi fecundabili. Gameţii rezultaţi nu sunt identici, fiecare are o altă combinaţie de gene datorită fenomenelor de crossing‑over şi segregare independentă a cromosomilor.
1.2. particularităţile GAMETOGENEZEI la bărbat şi femeie
Meioza prezintă o serie de particularităţi la cele două sexe, legate de cronologia, desfăşurarea şi finalitatea ei. La bărbat se formează în final patru spermatozoizi, în timp ce la femeie, prin eliminarea globulilor polari, rezultă doar un ovul.
a). Gametogeneza masculină
Spermatogeneza începe la pubertate (sub acţiunea FSH şi LH) şi continuă toată viaţa adultă. Fiecare spermatocit primar va produce prin meioza I două spermatocite secundare haploide, care după ce parcurg meioza II dau naştere la patru spermatide; ele se vor diferenţia în spermatozoizi, de două feluri: jumătate au un cromosom X, iar jumătate au un cromosom Y. Cromosomii X şi Y conţin fiecare gene de sexualizare specifice fiecărui sex. Aşa cum am văzut, în profaza meiozei I ei se unesc „cap la cap” (şi nu „margine la margine”, ca autosomii); această configuraţie împiedică schimbul de gene dintre X şi Y şi, deci, păstrarea unui singur tip de determinanţi sexuali, fie masculini, pe Y, fie feminini pe X. Acest fenomen, important în evoluţie, a permis realizarea gonocorismului (gonade distincte la indivizi diferiţi)
Întregul proces de spermatogeneză durează aproximativ 64 de zile, indiferent de vârstă; deci meioza masculină este un proces rapid iar bărbatul este totdeauna „tânăr” prin gameţii săi.
Meioza masculină este un proces intens, 1 ml de spermă conţinând în mod normal circa 70 de milioane de spermatozoizi. Ţinând cont de faptul că numai un singur spermatozoid este fecundant, am putea aprecia că gametogeneza masculină este „neeconomică” şi prin aceasta, lipsită de sens biologic („natura nu face risipă”). În realitate numărul enorm de spermatozoizi realizează o populaţie considerabilă în care şansele de realizare a unui fenomen sunt teoretic egale cu cele de realizare a fenomenului contrar. Aşadar, şansele unui spermatozoid cu X de a fecunda un ovul cu X şi a da un zigot XX sunt egale cu şansele unui spermatozoid cu Y de a se uni cu un ovul cu X şi de a forma un ou XY. În felul acesta proporţia sexelor la naştere este aproape 1:1.
Studiile efectuate asupra timpului şi ritmului desfăşurării meiozei masculine au evidenţiat că acest proces este autoreglabil, căci odată declanşat el se autoîntreţine ritmic, ca „o reacţie în lanţ”, nefiind condiţionat de factori externi; dar meioza masculină este sensibilă la acţiunea factorilor de mediu (poziţia intra-abdominală în criptorhidie, temperatura ridicată, ischemia) care pot reduce eficenţa spermatogenezei.
Desfăşurarea ritmică şi regulată a meiozei masculine asigură o deplină siguranţă procesului de distribuţie al cromosomilor. Creşterea vârstei tatălui nu produce o creştere a frecvenţei erorilor de distribuţie şi deci a anomaliilor cromosomice. În schimb, meioza masculină produce fără oprire cópii celulare identice (20-25 cicluri replicative pe an). Dacă apare o mutaţie genică aceasta este copiată neîntrerupt: la vârste tinere numărul de celule ce vor avea mutaţia este mic dar la vârste mai înaintate el devine semnificativ, producând un veritabil mozaic germinal. De aceea, odată cu creşterea vârstei, bărbaţii sănătoşi au un risc crescut şi recurent de a avea copii cu afecţiuni monogenice autosomal dominante (neurofibromatoză, acondroplazie, ş.a.) (vezi capitolul 5.D.6), deoarece o mutaţie apărută este copiată neîntrerupt.
Tabel 5.2 Diferenţe în gametogeneză la bărbat şi femeie
|
|
Bărbat |
Femeie |
|
Debut |
La pubertate |
În viaţa embrionară |
|
Desfăşurare |
Continuă |
Discontinuă |
|
Număr de mitoze în formarea gameţilor |
30-500 |
20-30 |
|
Reglare |
Autoreglabil |
Condiţionat de factori externi |
|
Număr de gameţi per meioză |
4 spermatide, de două feluri (cu X şi cu Y) |
1 ovul + 3 globule polare (numai cu X) |
|
Durata procesului |
Rapid: 64 zile |
Lent: 10-50 de ani |
|
Intensitate: număr de gameţi în viaţa adultă |
Intens: circa 100-200 milioane per ejaculare |
F.puţin intens: 1 ovul per ciclu menstrual |
|
Erori |
De copiere (risc de mutaţii genice) |
De distribuţie a materialului genetic (risc de anomalii cromosomice) |
b) Gametogeneza feminină
Spre deosebire de bărbat, la femeie ovogeneza începe prenatal şi este discontinuă: în luna a III-a de viaţă intrauterină ovogoniile înep să se dividă şi se transformă în ovocit primare; în luna VIII-a ovogoniile dispar aproape complet deoarece s-au diferenţiat în ovocite. Deci, spre deosebire de bărbat, la care spermatogoniile se produc (după pubertate) continuu până la bătrâneţe, „capitalul” de ovocite al ovarului este limitat (la naştere există circa 2 milioane de ovocite în fiecare ovar iar la pubertate mai putin de 200.000).
Ovocitele primare au deasemenea câteva particularităţi ce le deosebesc de spermatocite: sunt celule mari, au un nucleu excentric şi încep imediat meioza I (luna III-a) dar nu depăşesc stadiul de leptoten, fiind blocate într-o „fază de aşteptare” numită dictioten (prin intervenţia unei proteine numită OMI de la „oocyte meiosis inhibitors”); ele rămân în această fază mulţi ani, până la ovulaţie. După pubertate, sub acţiunea LH hipofizar, cîteva ovocite îşi încep lunar maturarea dar de regulă numai unul singur (maximum două) termină meioza I, formând două celule haploide inegale (datorită poziţiei excentrice a nucleului): ovocitul II (care preia aproape toată citoplasma ovocitului I) şi primul globul polar. Meioza II începe imediat dar este oprită în metafaza II şi nu se reia decît dacă s-a produs fecundarea. Atunci are loc expulzia celui de al doilea globul polar. Devine evident că meioza la femeie este un proces discontinuu şi condiţionat de factori externi (FSH şi LH, ovulaţia, fecundarea). La femeie, prin meioză, dintr-un ovocit I rezultă o singură celulă sexuală matură, aptă de fecundare (şi trei globule polare, care vor degenera). Toate ovocitele vor avea un cromosom X.
Ritmul lent al ovogenezei (un ovul pe lună) face ca gametogeneza feminină să fie puţin intensă: în cursul perioadei reproductive (de numai 30 ani, deoarece meioza feminină se opreşte definitiv la menopauză) se maturează numai 300-400 ovocite. Din acest motiv apar puţine „erori de copiere” (se produc copii puţine dar impecabile). Totuşi, ovogeneza nu este ferită de accidente: odată cu creşterea vârstei reproductive, mai ales după 35 de ani, se produc mai frecvent accidente de distribuţie a cromosomilor rezultând gameţi cu anomalii cromosomice de număr care, după fecundare, vor produce zigoţi anormali. Aceste erori sunt determinate de particularităţile meiozei la femeie şi în special de faptul că meioza este discontinuă, necesită intervenţia unor factori externi iar ovocitele…”au vârsta femeii”.
1.3. ACCIDENTE DE DISTRIBUŢIE A LE MATERIALULUI GENETIC
îN MEIOZĂ ŞI CONSECINŢELE LOR
În desfăşurarea meiozei se pot produce două categorii de erori, corespunzătoare celor două procese majore care au loc în acest proces: recombinarea genică şi segregarea (disjuncţia) anafazică a cromosomilor. Prima categorie este reprezentată de crossing-overul inegal iar cea de a doua de nedisjuncţia meiotică, pierderea unor cromosomi sau nesepararea citelor de ordinul II.
a. Erorile de recombinare
Fenomenul normal de recombinare genică produs în profaza MI (pahiten), prin crossing-over, presupune alinierea cromosomilor omologi, împerecherea secvenţelor omologe şi un schimb reciproc şi egal de material genetic. Uneori se poate produce accidental un crossing-over inegal, o recombinare „ilegitimă”, care are loc între secvenţe omologe de ADN (deseori repetitive) dar nealele (nu sunt situate în aceeaşi poziţie în cromosomi), care s-au aliniat (împerecheat) incorect în zigoten (figura 5.18); după crossing-over rezultă cromosomi cu duplicaţii sau deleţii ale unor segmente mici (ce conţin una sau mai multe gene).
Fenomenul de recombinare omologă nealelică (inegală sau ectopică) este considerat procesul fundamental al evoluţiei unor gene prin duplicaţie (de exemplu, evoluţia genelor globinelor; capitolul 3, figura 3.10) precum şi mecanismul creşterii numărului de secvenţe repetate în tandem, din structura ADN înalt repetitiv (vezi figura 6.8). CO inegal are, evident, şi consecinţe patologice deoarece duplicaţia sau deleţia unor mici regiuni cromosomice poate produce un fenotip anormal. Acest mecanism de producere a ”macroleziunilor din ADN” a fost identificat relativ recent, prin analiză moleculară, în mai multe boli (talasemia α, boala Charcot-Marie-Tooth, daltonismul, ş.a) precum şi în multe sindroame cu microdeleţie sau microduplicaţie (care alcătuiesc împreună grupul de boli genomice) (vezi capitolul 6.B.1.3.a).
Boala Charcot-Marie-Tooth (CMT) sau neuropatia motorie şi senzorială ereditară este cea mai frecventă boală neurologică ereditară (1:2500 nn), se caracterizează prin degenerarea progresivă a mielinei nervilor periferici şi consecutiv, hipotonie şi atrofie musculară (în special peonieră). Boala este heterogenă (3 tipuri) cel mai frecvent fiind tipul 1A, autosomal dominant, cu locusul lângă centromerul cromosomului 17 (în banda 17p11.2). CMT 1A este asociată cu o duplicaţie a unui segment de ADN de 1.5 Mb, ce conţine gena PMP22 ce codifică o proteină a mielinei periferice (figura 5.18). La fiecare capăt al genei se află o secvenţă nucleotidică identică, de circa 30 K.b, numită MITE[27]; prin aliniere incorectă şi CO inegal rezultă un cromosom cu duplicaţie genei care va da clinic CMT şi un cromosom cu deleţia genei care va da clinic o altă boală neurologică HNPP (de la hereditary neuropathy with liability to pressure palsies – neuropatie ereditară cu predispoziţie la pareze presionale) .
Sindroamele cu microdeleţii sau microduplicaţii (vezi capitolul 10.C.2) sunt sindroame dismorfice în care prin deleţia / duplicaţia unor segmente cromosomice mici (identificabile prin tehnici de înaltă rezoluţie sau FISH, vezi capitolul 2.D.3.a) se produce pierderea (haploinsuficienţa) sau trisomia parţială a unor gene multiple, contigue (vecine) (de unde şi denumirea sindroamele genelor contigue). Analiza moleculară fină a relevat că punctele de ruptură se localizează în secvenţele repetitive identice ce flanchează segmentul implicat şi că recombinarea anormală între copiile vecine ale secvenţele repetitive, produce deleţia sau duplicaţia regiunii respective (figura 5.19); un alt exemplu îl reprezintă sindromul velocardiofacial (deleţia 22q11.2), cu o frecvenţă de 1:4000 nn.
b. Erorile de distribuţie
În anafazele meiozei I şi II materialul genetic este împărţit egal şi total celulelor fiice. Acest proces poate suferi, ca şi în mitoză, erori de distribuţie (de segregare) care vor duce la formarea unor gameţi neechilibraţi genetic; după fecundarea lor vor rezulta zigoţi anormali. În funcţie de evoluţia acestor zigoţi se pot produce avorturi spontane, nou-născuţi morţi sau nou-născuţi vii plurimalformaţi. Mecanismele principale de apariţie a acestor erori sunt: nedisjuncţia (cromosomică, cromatidiană), pierderea unor cromosomi (ca urmare a migrării întârziate în anafază), nesepararea citelor de ordinul II (celule rezultate după meioza II).
(1). Nedisjuncţia este lipsa separării cromosomilor ce formează o pereche de omologi, în anafaza meiozei I (nedisjuncţie cromosomică) sau a cromatidelor surori ale unui cromosom (nedisjuncţie cromatidiană) în meioza II; datorită acestei erori, ambele „unităţi” (cromosomul sau cromatida) trec într-o celulă fiică şi vor lipsi în cealaltă celulă. Prin nedisjuncţie în meioza I sau II rezultă gameţi disomici şi nulisomici care, după fecundarea cu un gamet normal, vor forma un zigot trisomic sau monosomic (vezi capitolul 6.B.1). Deşi dezechilibrul cromosomic produs de cele două tipuri de nedisjuncţia este de acelaşi tip, între nedisjuncţia din meioza I şi nedisjuncţia din meioza II sunt totuşi diferenţe mai subtile (figura 5.20). Dacă eroarea are loc în meioza I, toţi gameţii vor fi anormali iar gametul disomic, cu 24 de cromosomi, va avea ambii cromosomi (matern sau patern) ai perchii; dacă nedisjuncţia are loc în meioza II numai jumătate din gameţi vor fi anormali iar gametul disomic va avea ambele copii (cromatide) fie ale cromosomului matern, fie ale celui patern (disomie uniparentală).[28] (Caseta 5.3)
(2). Întârzierea anafazică constă în pierderea unui cromosom în cursul anafazei meiozei I sau II. În consecinţă, apar gameţi nulisomici (n‑1) care prin fecundare, vor forma zigoţi monosomici (2n‑1); majoritatea sunt incompatibili cu supravieţuirea (excepţie 45, X)
(3). Nesepararea "citelor" de ordin II este un eveniment prin care se formează gameţi diploizi, care au întregul set de cromosomi (2n). Prin fecundare cu gameţi normali apar zigoţi triploizi, neviabili (3n = 69 cromosomi).
Caseta 5.3
NEDISJUNCŢIA
Nedisjuncţia reprezintă o eroare de segregare (separare) a cromosomilor în anafaza meiozei sau mitozei. În nedisjuncţia meiotică se pot produce gameţi anormali (disomici şi nulisomici) care, după fecundare, vor da zigoţi cu anomalii cromosomice numerice. In nedisjuncţia cromatidiana se formează (în diferite momente ale dezvoltării) clone celulare anormale, ce vor evolua alături de celulele normale, formând un mozaic cromosomic. Trebuie să precizăm că există două tipuri de erori de separare: (1) nesepararea cromosomilor omologi (meioza I) sau a cromatidelor surori (meioza II) (figura 5.20); (2) separarea prematură a cromatidelor surori în meioza I în loc de meioza II. Ambele tipuri de erori produc gameţi anormali.
Nedisjuncţia este un fenomen „tulburător” atât din punct de vedere teoretic cât şi din punct de vedere practic. „Tulburător” pentru că ridică câteva întrebări importante dar dificile: este frecvent? se produce la fel de des în meioza maternă şi paternă? care sunt cauzele nedisjuncţiei? poate fi evitată? care este riscul de nedisjuncţie şi deci de naştere a unor feţi cu anomalii cromosomice de număr?
La prima întrebare răspunsul este afirmativ: nedisjuncţia este frecventă, mai ales în meioză şi în mod special în meioza maternă. Se estimează că 8% din toate concepţiile (zigoţii) umane au anomalii cromosomice; majoritatea sunt neviabile şi se elimină prenatal, astfel că numai 0,7-1% din nou născuţii vii au o anomalie cromosomică. În toate cazurile predomină anomaliile numerice (aproape 90%), produse în special de nedisjuncţie.
Originea nedisjuncţiei, maternă sau paternă, se poate stabili în prezent relativ uşor cu ajutorul unor markeri ADN polimorfici. Toate studiile demonstrează că nedisjuncţia este mult mai frecventă în ovogeneză, în special în meioza I. Dacă ne referim numai la trisomia 21, cea mai frecventă boală cromosomică (1:700 nn), s-a stabilit (Antonarkis,1993) că în 92% din cazuri sindromul Down este produs prin nedisjuncţie maternă în meioza I, în 5% din cazuri prin nedisjuncţie paternă în meioza II şi în 3% din cazuri prin nedisjuncţie în mitozele postzigotice. La aceste date clare se adaugă un alt lucru cert: riscul nedisjuncţiei creşte odată cu creşterea vârstei reproductive materne, mai ales după 35 de ani. Statisticile, pe loturi mari, sunt foarte detaliate stabilind un risc precis pentru fiecare an de vârstă; dar pentru a ilustra fenomenul este suficient să precizăm că la gravidele de 30 de ani riscul naşterii unui copil cu sindrom Down este de 1:1000; el creşte la 1:100 la 37 de ani şi la 1:10 peste 45 de ani (Gardner şi Sutherland, 1996). Nedisjuncţia maternă este principala „cauză” a sindromului Down şi ale altor trisomii autosomale (după Hassold, 1996 peste 50% din ovocitele ovulate la femei de 40-45 de ani pot fi aneuploide). Sunt totuşi situaţii în care nedisjuncţia meiotică paternă este regula (de exemplu trisomia XYY) sau este relativ frecventă (70% din sindromul Turner, 45,X).
Care sunt cauzele nedisjuncţiei meiotice? Aici există trei certitudini dar care …nu rezolvă problema, deşi sunt utile în practica sfatului genetic.
Efectul vârstei materne (ca factor predispozant la nedisjuncţie) este sigur şi foarte probabil se corelează cu particularităţile meiozei materne[29], care (printre altele) blochează ovocitele timp de câteva decade în profaza meiozei I („ovulele au vârsta femeii”). Aceasta duce la o recombinare meiotică aberantă (sinapse incorecte, schimburile intercromosomice cu o rată redusă şi o localizare anormală) şi la anomalii în formarea fusului de diviziune. Nu trebuie însă să uităm că numai un sfert din cazurile de sindrom Down se nasc din femei peste 35 de ani; deci nedisjuncţia maternă are probabil şi alte cauze decât vârsta maternă reproductivă avansată, care, de ce nu, ar putea acţiona şi la femeile peste 35 de ani. Si mai trebuie să spunem clar un lucru: la 40 de ani o gravidă are doar un risc de 1-2% de a naşte un copil cu sindrom Down (este drept, mult mai mare decât la 25 de ani), deci un risc relativ mic şi acceptabil, mai ales în condiţiile existenţei diagnosticului prenatal;
În ciuda cercetărilor intense, nu există nici-o dovadă ştiinţifică care să susţină rolul posibil al unor factori externi: infecţii virale, radiaţii, medicamente, hormoni, contraceptive, cafeaua sau fumatul, reducerea frecvenţei raporturilor sexuale etc. De aceea în practica sfatului genetic rolul invocat de altii al acestor factori trebuie cel putin…temperat.
La om, nu există probabil factori genetici care să crească riscul de nedisjuncţie); pentru aceasta pledează riscul de recurenţă mic (1%) pentru un al doilea copil afectat (atenţie: la acest risc trebuie adăugat riscul dependent de vârsta maternă). Totuşi nu trebuie să uităm că la alte specii nedisjuncţia este sub control genetic iar diferiţi cromosomi au rate diferite de trisomie în avorturile spontane (de exemplu, trisomia 16 reprezintă o treime din aceste avorturi). Oricum, controlul genetic al meiozei este încă departe de a fi elucidat
FECUNDAREA
Prin fecundare se înţelege unirea celor doi gameţi, ovulul şi spermatozoidul, şi formarea zigotului (celula ou), din care se va dezvolta un nou organism. În felul acesta, părinţii transmit, prin gameţi, „zestrea” lor ereditară viitorilor descendenţi.
Fecundarea este un fenomen complex, cu anumite caracteristici; dintre acestea vom reţine, pentru cadrul discuţiei noastre, faptul că la om fecundarea este monospermică: pătrunderea unui singur spermatozoid în ovul determină o serie de evenimente biochimice ce împiedică intrarea altor spermatozoizi. Concomitent, aparatul mitotic al ovocitului (situat la periferie) este brusc activat şi se finalizează meioza II, eliminându-se al doilea globul polar. Setul de cromosomi ce ramâne în ovocit formează pronucleul feminin. În citoplasma ovulului, spermatozoidul suferă o serie de modificări (coada şi piesa intermediară, ce are câteva mitocondrii, sunt dezintegrate) iar nucleul său devine pronucleul masculin. Cei doi pronuclei se aproprie unul de altul, cromosomii lor se replică sincron şi, după ruperea membranelor nucleare, se fixează pe fibrele primului fus de diviziune; zigotul se divide şi formează primele două celule fiice diploide.
În cursul fecundării au loc mai multe evenimente genetice. În primul rând, prin fuziunea celor doi gameţi haploizi (n=23) se reface numărul diploid de cromosomi, caracteristic speciei umane (2n=46). Începând cu zigotul toate celulele somatice vor avea cromosomii în perechi de omologi, identici ca morfologie şi structură genetică dar diferiţi ca origine. În momentul fecundării, genele parentale din cromosomii gameţilor formează o structură nouă, total diferită de a părinţilor, unică şi constantă; ea determină individualitatea genetică a organismului. Contribuţia părinţilor la ereditatea copiilor nu este perfect egală deoarece ADN mitocondrial provine exclusiv de la mama, prin mitocondriile din citoplasma ovulului.
În al doilea rând, în momentul fecundării se stabileşte sexul genetic XX sau XY al viitorului organism, prin asortarea cromosomilor sexuali din gameţi. Întrucât la mamifere şi om sexul masculin este heterogametic, el va produce două tipuri de spermatozoizi cu X şi cu Y, care fecundând un ovul cu Y vor realiza sexul genetic XX şi respectiv XY. Aşa cum am arătat, numărul foarte mare de spermatozoizi lansat în cursul unui raport sexual fecundant asigură condiţiile statistice care oferă celor două tipuri de spermatozoizi şanse egale de fecundare; de aceea raportul sexelor la naştere este aproape 1:1.
În realitate, studiile sexului genetic la fetuşi au arătat o uşoară predominenţă a sexului masculin (120 fetuşi masculini la 100 feminini, deci 54,5%); o explicaţie posibilă ar fi mobilitatea mai mare a spermatozoizilor cu Y, care ajung mai frecvent la „ţintă”. Diferenţele se mai reduc la naştere (51,5%) şi se pare că se anulează la pubertate, pentru ca în a treia decadă de viaţă raportul să fie favorabil sexului feminin. Această „dinamică” nu este clar explicată; o ipoteză susţine că sexul feminin este „mai rezistent” la agresiunile externe deoarece are un cromosom X în plus, care nu ar fi total inactivat (conform ipotezei Lyon) (vezi capitolele 2.D.2.2 şi 4.B.4.a).
În cursul fecundării se pot produce o serie de devieri de la mecanismul sau rezultatul procesului normal de fecundare. Există astfel posibilitatea rară a unei duble fecundări atunci când ovarul eliberează în momentul ovulaţiei două ovule. Prin fecundarea lor individuală se vor forma doi gameţi; ei pot evolua separat, independent unul de altul, formând gemenii dizigoţi sau se pot uni într-o singură masă embrionară ce va produce un singur individ cu două componente genetice distincte ca origine („doi indivizi într-unul singur”), numit himeră (după monstrul din mitologia greacă care avea cap de leu, corp de capră şi coadă de balaur). În ultimul caz, dacă zigoţii vor avea acelaşi sex genetic se realizează o sexualizare normală şi numai studiul unor caractere ereditare normale (grupe sanguine, serice etc) va evidenţia existenţa unei duble populaţii celulare[30]. Dacă zigoţii care s-au unit aveau sexe genetice diferite, se realizează o constituţie genetică XX/XY care va produce o tulburare de sexualizare, numită hermafroditism adevărat.
O altă anomalie de fecundare este dispermia, situaţia foarte rară în care un ovul este fecundat de către doi spermatozoizi. Rezultă un zigot triploid (3n). Acelaşi rezultat poate fi obţinut atunci când unul din gameţi este diploid (diginie sau diandrie) (vezi capitolul 6.B.1). Evoluţia unui zigot triploid va depinde de originea setului haploid suplimentar: dacă este de provenienţă maternă (diginie) atunci dezvoltarea embrionară este sever întârziată, placenta este mică şi fibrotică iar embrionul este avortat precoce; dacă este de origine paternă (diandrie) se va forma o placentă anormală, mare şi polichistică, cu un embrion slab dezvoltat (molă hidatiformă parţială)
Vom menţiona în sfârşit situaţia în care un spermatozoid 23,X fecundează un ovul fără nucleu şi setul de cromosomi al spermatozoidului se dublează formînd un zigot 46,XX în care toţi cromosomii sunt de origine paternă şi toate genele alele vor fi homozigote; se produce o dezvoltare anormală a trofoblastului şi dezorganizarea/absenţa embrionului numită mola hidatiformă completă. Există şi situaţia inversă, în care celule 46,XX conţin numai cromosomi materni; ele proliferează şi produc un teratom ovarian (alcătuit din celule embrionare dar nu şi din ţesut placentar). Din cele două situaţii prezentate mai sus rezultă că dezvoltarea fetală normală necesită ambele contribuţii genetice, maternă şi paternă.
D. EREDITATEA MONOGENICĂ
1. Legile lui Mendel.
Transmiterea caracterelor ereditare de la părinţi la copii a fost remarcată din cele mai vechi timpuri. Dar explicaţiile date similitudinii familiale şi încercările de a stabili legile eredităţii au cunoscut numeroase eşecuri. Ele erau generate de ipoteza greşită a “amestecării caracterelor ereditare”. Potrivit aceastei ipoteze, descendenţii prezintă un amestec al caracterelor parentale; ele îşi pierd identitatea şi nu se vor mai regăsi ca atare în generaţiile următoare.
Pe baza unor cercetări experimentale de mare fineţe şi precizie, Gr. Mendel a demonstrat (1865) că la urmaşi nu se produce nici un amestec al caracterelor parentale; unele caractere nu se exprimă în prima generaţie filială dar pot apărea neschimbate ulterior. Pentru a explica aceste fenomene, Mendel a presupus că fiecare caracter este determinat de o pereche de factori ereditari (numiţi mai târziu „gene”), care în momentul formării gameţilor se separă, segregă, distribuindu-se la întâmplare în celulele sexuale. Mendel a observat că transmiterea caracterelor ereditare este un proces statistic ce poate fi descris în termeni de probabilitate, şi a formulat legile care guvernează acest proces: legea segregării şi legea asortării independente; ele stau la baza geneticii ca ştiinţă.
Cercetările şi rezultatele obţinute de Mendel au fost ignorate de către contemporanii săi “nepregătiţi” pentru a le înţelege. În anul 1900 principiile stabilite de Mendel au fost redescoperite simultan în trei laboratoare, demonstrându-se că legile eredităţii ar fi fost descoperite, la un anumit nivel al dezvoltării cunoştinţelor, chiar şi….în absenţa lui Mendel, deoarece legile eredităţii sunt legi obiective. Doi ani mai târziu, Sutton, pornind de la ipoteza (plauzibilă) că materialul ereditar se găseşte în cromosomi, stabileşte o corelaţie strânsă între comportamentul cromosomilor în meioză şi principiile enunţate de Mendel, formulând “teoria cromosomică a eredităţii”. În acelaşi an (1902), un medic englez, sir A.Garrod, a raportat primul exemplu de ereditate mendeliană la om – alcaptonuria – o anomalie biochimică congenitală, transmisă ereditar, recesiv. Ulterior el a studiat şi alte boli ereditare (albinismul, cistinuria şi pentozuria), veritabile “erori înnăscute de metabolism”, şi a pus astfel bazele geneticii medicale.
Datele acumulate de genetica secolului XX au demonstrat universalitatea legilor lui Mendel. Numeroase caractere umane normale sau anormale, determinate monogenic, se transmit în concordanţă cu aceste legi. „Dacă Mendel ar fi trăit în epoca noastră, i-ar fi fost probabil mult mai uşor să descopere legile geneticii studiind ereditatea familiilor umane” (Ch. Salmon, 1973).
Dezvoltarea geneticii moleculare a modificat în mare măsură “mendelismul clasic” mai ales prin înţelegerea modului de exprimare a genei şi a interacţiunilor complexe dintre gene. “Nu este vorba de o răsturnare a principiilor care guvernează transmiterea mendeliană, ci numai de o dezvoltare a lor” (Hull, 1978).
1.1. PRIMA LEGE A LUI MENDEL: LEGEA SEGREGĂRII
Prima lege a lui Mendel sau legea segregării[31] a fost stabilită şi poate fi demonstrată prin urmărirea rezultatelor încrucişărilor între indivizi care diferă printr-un singur caracter (monohibridare), sau – mai exact – printr-un cuplu de caractere alternante sau contrastante.
Vom analiza un exemplu concret. După capacitatea de a simţi gustul amar al feniltiocarbamidei (PTC) sau al feniltioureei, circa 70% din populaţie este alcătuită de “gustători” şi 30% din “negustători”. Din căsătoria unui gustător cu un negustător (generaţia P sau parentală) rezultă în F1 (prima generaţie filială) numai descendenţi gustători (100%); încrucişarea lor va produce în F2 atât gustători cât şi negustători. Caracterul parental gustător care se manifestă în F1 şi F2 este dominant iar cel negustător, care apare numai în F2, se numeşte recesiv. Cumulând mai multe observaţii familiale şi calculând proporţia celor două fenotipuri, se obţine în F2 un rezultat egal sau vecin cu 3:1.
Persoanele negustătoare din F2, prin încrucişare cu indivizi asemănători lor, vor da numai descendenţi negustători. În schimb, din încrucişarea gustătorilor din F2 cu negustători[32] rezultă, în circa 1/3 din cazuri, numai gustători iar în restul de 2/3 apar în proporţii egale gustători şi negustători; acest lucru ne arată că deşi fenotipic gustătorii din F2 sunt identici, genotipic ei sunt diferiţi. Ţinând cont de această diferenţă şi de raporturile procentuale, rezultă că indivizii din F2 aparţin fenotipic la două categorii, în proporţie de 3 : 1, iar genotipic ei alcătuiesc trei grupe, în raportul de 1 : 2 : 1.
Pe baza acestui exemplu se poate realiza un model general valabil al transmiterii caracterelor în monohibridări. El va fi interpretat în spiritul gândirii mendeliene, folosind însă noţiunile şi cunoştinţele acumulate ulterior de genetica clasică şi în special cele despre cromosomi şi dinamica lor în meioză.
Mendel a presupus că orice individ rezultat prin unirea celor două tipuri de gameţi parentali primeşte material genetic de la ambii părinţi. De aceea, fiecare caracter este determinat de o pereche de “factori ereditari” (termenul îi aparţine lui Mendel) sau gene alele (ce ocupă acelaşi locus pe cromosomii omologi). Organismele din generaţia parentală (P) sunt “pure” din punct de vedere genetic, au genele alele identice, sunt homozigote: A/A şi a/a. În anafaza meiozei I genele alele segregă şi gameţii (haploizi) conţin un singur “factor ereditar” sau genă: (A) şi (a); deci homozigoţii sunt homogametici. Prin fecundarea acestor gameţi rezultă un zigot (diploid) în care factorii ereditari materni şi paterni se asociază alcătuind genotipul A/a al tuturor descendenţilor hibrizi din F1. Ambii părinţi contribuie în mod egal la ereditarea copiilor (tabelul 5.3).
Tabelul 5.3 Monohibridarea şi legea I a lui Mendel
|
Generaţie |
Fenotipuri |
Genotipuri |
|
|
P
F1 |
A x a
A (100%) |
A/A x a/a (A) (a)
A/a |
homozigoţi gameţi
heterozigoţi |
|
Încrucişare indivizi F1
F2
|
A x A
A şi a (75%) (25%) |
A/a x A/a (A) şi (a) (A) şi (a) A/A A/a a/a ( 25% 50% ) 25% |
heterozigoţi gameţi
|
|
Retroîncrucişare F1 x P(recesiv) |
A(F1) x a
A şi a (50%) (50%) |
A/a x a/a (A) şi (a) (a)
A/a a/a
|
gameţi
|
|
Retroîncrucişare F2 x P(recesiv) |
(1/3) A(F2) x a
A (100%) |
A/A x a/a (a)
A/a
|
gameţi |
|
(2/3) A(F2) x a
A şi a (50%) (50%) |
A/a x a/a (A) şi (a) (a)
A/a a/a
|
gameţi |
Descendenţii din F1 au gene alele diferite, sunt heterozigoţi. Întrucât au un genotip identic (A/a), toţi indivizii (hibrizi) din prima generaţie filială sunt fenotipic identici, uniformi. Aceasta este o regulă generală denumită “principiul uniformităţii hibrizilor din F1” (după unii este “prima” lege a lui Mendel): când se încrucişează indivizi homozigoţi (“puri”) ce diferă printr-un caracter particular, descendenţii lor (F1) sunt uniformi.
Să analizăm acum încrucişările dintre indivizii din generaţia F1. În F2 se obţin fenotipic două tipuri de urmaşi: unii dintre ei se aseamănă cu fenotipul dominant din P şi F1, iar alţii cu fenotipul recesiv din P. Proporţia lor este de 3 dominanţi la 1 recesiv. Se observă că în F2, alături de caracterul dominant reapare caracterul parental recesiv, care nu s-a exprimat în prima generaţie filială, deşi gena corespunzătoare (a) era prezentă (A/a). Rezultă că factorii ereditari sau genele care determină caracterul recesiv îşi menţin neschimbată identitatea lor, chiar dacă nu se manifestă fenotipic în F1. Ele nu se “contaminează”, nu „se amestecă” (aşa cum se credea înainte de Mendel), rămân neschimbate, constante şi riguros “pure”. Gena recesivă devine manifestă în momentul în care (în F2) se realizează starea homozigotă (a/a). “Un caracter recesiv circulă de-a lungul generaţiilor ca un râu subteran, care reapare din timp în timp, atunci când se produce confluenţa a două din braţele sale” (Ph. Sentis).
Prin studiul descendenţilor F3 (sau prin retroîncrucişare F2 x Precesiv) s-a demonstrat că în F2 au rezultat în realitate trei tipuri de descendenţi: ¼ seamănă cu părintele dominant, ½ sunt hibrizi ca şi indivizii din F1 şi ¼ erau puri şi asemănători părintelui recesiv. Acest raport genotipic 1:2:1 în F2 indică faptul că jumătate din gamaţii unei persoane F1 poartă gena dominantă iar cealaltă jumătate – pe cea recesivă. Această concluzie a fost capitală pentru Mendel, căci ea i-a permis să înţeleagă mecanismul transmiterii genetice şi să formuleze legea segregării:
orice individ primeşte, în mod egal, material genetic (gene) de la ambii părinţi;
genele parentale se separă (segregă) atunci când acel individ produce la rândul său gameţi;
gameţii vor fi “puri” genetic (au o singură genă din perechea de alele);
prin combinarea întâmplătoare a gameţilor, în F2 se produce o separare, o disjuncţie, a caracterelor parentale, în proporţia de 3 dominanţi la 1 recesiv.
Legea segregării se explică astăzi perfect prin dinamica cromosomilor omologi în meioza primară (tabelul 5.3): ei se separă (disjuncţie) şi merg în celule distincte. Heterozigoţii (A/a) din F1 produc două tipuri de gameţi (A) şi (a). Fiecare genă alelă din genotipul A/a are o probabilitate egală de a se transmite şi de a se găsi în gameţi, indiferent dacă este dominantă sau recesivă. Cele două tipuri de gameţi se produc în cantităţi statistic egale. Fiecare gamet are, de asemenea, o probabilitate egală de a se combina (fecunda) cu ceilalţi. Prin întâlnirea lor întâmplătoare pot rezulta în F2 trei tipuri de indivizi cu genotipurile A/A, A/a, a/a în proporţie de 1 : 2 : 1. În funcţie de raporturile de dominanţă /recesivitate dintre gene vor rezulta proporţiile fenotipice de 3 dominaţi : 1 recesiv.
Menţionăm, în finalul acestei discuţii, că acţiunea genei este independentă de originea sa maternă sau paternă. Când se formează un zigot, genele “par a uita” originea lor parentală.
1.2. A DOUA LEGE A LUI MENDEL: LEGEA ASORTĂRII INDEPENDENTE
A doua lege a lui Mendel sau legea asortării independente[33] poate fi demonstrată prin încrucişări între organisme ce se deosebesc prin două (dihibridare) sau mai multe cupluri de caractere. Ele se transmit independent unul de altul: ereditatea primului caracter nu influenţează ereditatea celui de al doilea. Explicaţia acestui fenomen rezidă în faptul că genele ce determină aceste caractere sunt neînlănţuite, situate pe cromosomi omologi diferiţi si independente una faţă de alta; ele se pot combina liber între ele, dând genotipuri şi fenotipuri noi.
De exemplu: încrucişarea dintre o persoană “gustător şi cu grup sanguin A” cu alta “negustător şi cu grup sanguin 0”, va produce în generaţia F1 numai indivizi “gustători, cu grup sanguin A” (caractere dominante) (tabelul 5.4). Din încrucişarea lor rezultă în F2 patru tipuri de descendenţi: gustători – grup A; gustători – grup 0; negustători – grup A; negustători – grup 0, în proporţie de 9 : 3 : 3 : 1. Prima remarcă pe care o putem face în acest caz se referă la reapariţia în F2 a tipului parental recesiv: negustător – grup 0: Se remarcă apoi că în F2 apar, alături de fenotipurile parentale, două fenotipuri noi ce rezultă prin recombinarea caracterelor din generaţia P. Explicaţia acestor fenomene este astăzi simplă, întrucât cunoaştem structura şi funcţia genelor, precum şi dinamica cromosomilor în meioză (anafaza I).
Tabelul 5.4. Dihibridarea şi legea a II-a a lui Mendel
|
GENERAŢIE P fenotip: GUSTĂTOR; Grup sanguin A x negustător; grup sanguin 0 genotip: G/G ; A/A g/g; o/o gameţi: (G ; A) x (g ; o) F1 GUSTĂTORI; GRUP SANGUIN A. (100%)
|
F1 x F1 fenotip: GUSTĂTOR ; GRUP SANGUIN A x GUSTĂTOR ; GRUP SANGUIN 0
genotip: G/g ; A/o G/g ; A/o
|
Combinaţii fenotipice |
gameţi |
G ; A |
G ; o |
G ; A |
G ; o |
|
G ; A |
G/G ; A/A |
G/G ; A/o |
G/g ; A/A |
G/g ; A/o |
|
|
G ; 0 |
G/G ; A/o |
G/G ; o/o |
G/g ; A/o |
G/g ; o/o |
|
|
g ; A |
G/g ; A/A |
G/g ; A/o |
g/g ; A/A |
g/g ; A/o |
|
|
g ; 0 |
G/g ; A/o |
G/g ; o/o |
g/g ; A/o |
g/g ; o/o |

|
|
GUSTĂTORI |
|
Negustători |
|
|
GRUP SANGUIN A |
9 |
|
3 |
12 |
|
|
|
|
|
|
|
grup sanguin 0 |
3 |
|
1 |
4 |
|
|
12 |
|
4 |
|
Organismele parentale diferă prin două caractere distincte, determinate de gene situate pe cromosomi diferiţi (neînlănţuite). Aceste organisme sunt homozigote, întrucât genele alele sunt identice: (G/G; A/A) şi (g/g şi 0/0). Ele vor forma fiecare un singur tip de gameţi (G;A) şi (g;o), care – după fecundare – vor da prima generaţie filială (F1) de indivizi “uniformi” fenotipic şi dublu heterozigoţi (G/g; A/o); caracterul “gustător – grup A” este dominant. Prin încrucişarea acestor persoane F1 între ele, rezultă patru fenotipuri diferite, deoarece în momentul în care se formează gameţii, genele ce alcătuiesc cele două perechi de alele (G/g) şi (A/o), segregă (se separă) şi se asortează independent una de alta, întocmai ca şi cromosomii omologi în anafaza meiozei primare. Fiecare persoană F1 formează patru tipuri de gameţi: GA; Go; gA; go – care, prin fecundare întâmplătoare, vor da 16 genotipuri (de 9 feluri) (tabelul 5.4). Deoarece genele G şi A sunt dominante faţă de g şi o, rezultă numai patru categorii fenotipice parentale, în proporţie de 9:3:3:1; două sunt identice cu fenotipurile parentale, celelalte două sunt noi şi rezultă prin recombinarea lor. Apariţia acestor combinaţii noi pledează pentru faptul că asortarea genelor în gameţi este un proces în întregime întâmplător. Remarcăm faptul că fiecare caracter, luat separat, segregă independent de celălalt, realizând raportul specific pentru monohibridare de 3 : 1 (12 gustători : 4 negustători şi 12 grup A : 4 grup 0).
Legea a doua a lui Mendel sau legea asortării independente (sau a liberei combinaţii a factorilor ereditari) afirmă faptul că:
genele ce determină un caracter segregă independent de genele ce determină alt caracter;
genele ce alcătuiesc diferite perechi de alele (A/a; B/b; ş.a.m.d.) se asortează (se combină) independent una de alta, atunci când se formează gameţi;
în cazul a două perechi de caractere, raportul de segregare fenotipică în F2 este 9 : 3 : 3 : 1
La baza acestei legi stă fenomenul de asortare independentă a cromosomilor din perechile de omologi, ce are loc în cursul anafazei meiozei primare (recombinare cromosomică).
Legea a doua a lui Mendel poate fi demonstrată şi în cazul încrucişărilor între indivizi ce diferă prin 3, 4 … n cupluri de alele independente, distincte. Numărul şi diversitatea genotipurilor şi fenotipurilor care rezultă (admiţând că există o dominantă totală a unei alele asupra celeilalte) poate fi calculat conform tabelului 5.5
Tabelul 5.5 Diversitatea fenotipurilor şi genotipurilor ce rezultă din încrucişările între heterozigoţi pentru 1, 2, 3 … n cupluri de alele independente (după Bocque, 1974)
|
Număr de cupluri alele independente |
Număr de categorii genotipice |
Proporţii a diferitelor fenotipuri |
Număr de categorii genotipice |
|
1 (monohibridere) 2 (hibridare) 3 (trihibridare) 4 (tetrahibridare) |
21 = 2 22 = 4 23 = 8 24 = 16 |
(3:1)1 (3:1)2 (3:1)3 (3:1)4 |
31 = 3 32 = 9 33 = 27 34 = 81 |
|
n |
2n |
(3:1)n |
3n |
Numărul tipurilor de gameţi formaţi de fiecare sex este egal cu numărul categoriilor fenotipice indicate. La om, pentru n = 23, descendenţa posibilă între heterozigoţi numai pentru o singură pereche de alele pe fiecare din cele 23 perechi cromosomi ar fi 223 = 8 398 608 categorii fenotipice şi 323 = 94 143 178 827 categorii genotipice. Acest număr enorm de combinaţii teoretic posibile la descenţi, ne arată de ce fiinţele umane sunt practic unice, genotipic şi fenotipic.
2. TRANSMITEREA CARACTERELOR MONOGENICE
2.1. DATE GENERALE
Caracterele monogenice sunt cele mai simple caractere genetice a căror prezenţă sau absenţă depinde de alelele, normale sau mutante, ale unui singur locus, situat pe autosomi sau pe cromosomul X. Desigur, manifestarea caracterului poate depinde şi de alţi factori, genetici sau de mediu, dar genele respective sunt necesare şi suficiente pentru exprimarea caracterului. Ele au efecte mari şi se conformează în mod obişnuit principiului “o genă → o proteină → un caracter”, deşi, după cum ştim deja, mutaţii diferite în aceeaşi genă pot produce fenotipuri (boli) diferite sau, invers, mutaţii în gene diferite se pot manifesta fenotipic identic (vezi heterogenitatea genetică, capitolul 4.A). Aceste caractere monogenice se moştenesc de obicei într-un mod simplu, identic sau similar cu cel descris de către Mendel; de aceea ele se numesc şi caractere mendeliene.
Se cunosc peste 10.000 de caractere mendeliene, normale sau patologice, listate în lucrarea de referinţă a lui Victor McKusick Mendelian Inheritance of Man (ed.12, 1998), indispensabilă medicilor geneticieni. Versiunea online, numită OMIM este continuu actualizată şi disponibilă prin internet (caseta 5.4)
CASETA 5.4
OMIM
Online Mendelian Inheritance in Man (OMIM) este un catalog al genelor şi bolilor genetice care se transmit mendelian, accesibil prin Internet la adresa de web: http://www.ncbi.nlm.nih.gov/entrez/OMIM. OMIM este bazat pe textul celebrului catalog “Mendelian Inheritance in Man” care a fost elaborat în 1966 sub conducerea profesorului Victor McKusick de la Universitatea Johns Hopkins din Baltimore şi se află în prezent la ediţia a 12-a (1998). Spre deosebire de forma publicată care apare periodic, varianta online este actualizată zilnic. Astfel, la 1Martie 2003, numărul total de intrări era de 14231, număr care include atât fenotipuri pentru care sunt identificate genele cauzatoare, cât şi descripţii fenotipice fără loci identificaţi sau loci fără fenotip asociat.
Numărul de intrări OMIM la 21 februarie 2003
|
|
Autosomale |
Legate de X |
Legate de Y |
Mitocondriale |
Total |
|
Gene localizate sau fenotipuri cu loci precizaţi (prefix *) |
9956 |
541 |
41 |
37 |
10574 |
|
Fenotipuri cu doi sau mai multi loci asociaţi (prefix #) |
1120 |
94 |
0 |
23 |
1237 |
|
Alţi loci sau fenotipuri (fără prefix) |
2260 |
158 |
2 |
0 |
2419 |
|
Total |
13335 |
793 |
43 |
60 |
14231 |
* Un asterisc prezent înaintea unei intrări semnifică un fenotip determinat de gena situată la locusul precizat şi faptul că modul de transmitere a fenotipului a fost demonstrat, după aprecierea autorilor şi editorilor.
# Acest simbol înaintea unei intrări înseamnă că acest fenotip poate fi cauzat de mutaţii la nivelul a două sau mai multe gene.
Fiecare intrare are un numar unic din şase cifre. Prima cifră corespunde modului de transmitere a genei asociate: 1 – boli (sau loci) cu transmitere autosomal dominantă desemnate înainte de 15 mai 1994; 2 – boli (sau loci) cu transmitere autosomal recesivă stabilite înainte de 1994; 3 – boli (sau loci) legate de X; 4 – boli (sau loci) legate de Y; 5 – boli (sau loci) mitocondriale; 6 – boli (sau loci) autosomale desemnate după 15 mai 1994. Dupa cum se observă, după 1994 s-a renunţat la clasificarea genelor autosomale în dominante sau recesive deoarece unele gene pot avea mutaţii diferite cu transmiteri diferite. Variantele alelice ale unui locus sunt desemnate prin numere zecimale alcătuite din 4 cifre care urmează dupa codul de şase cifre. De exemplu, variantele alelice (mutaţiile) genei pentru factorul IX al coagulării sunt desemnate prin 306900.0001 până la 306900.0101. Locusul pentru beta-globina (HBB) este numerotat 141900, iar hemoglobina S (implicata in producerea sicklemiei) este numerotata 141900.0243.
Bolile monogenice sunt caracterizate prin modul lor de transmitere în familie, stabilit în urma unei anamneze (istoric) familiale şi reprezentat grafic (cu ajutorul unor simboluri standard), sub forma unui arbore genealogic (în engleză pedigree[34]) (figura 5.21).
Asupra acestei acţiuni, importante în diagnosticul bolilor genetice, ne vom referi cu detalii mai târziu (vezi capitolul 9.A), dar acum vom introduce câţiva termeni de bază, pentru a facilita discuţiile privind transmiterea genealogică. Persoana afectată (■ sau ● ) de la care începe recenzarea unei familii cu o boală genetică se numeşte proband (caz index sau propositus/proposita, în funcţie de sex) iar aceea care consultă medicul (rudă sănătoasă sau afectată cu probandul) se numeşte consultand. Relaţiile intrafamiliale şi gradele de rudenie sunt redate în figura 5.22. Poziţia unei persoane în arbore este indicată prin numere romane pentru generaţie şi numere arabe pentru locul unui individ într-o generaţie.
Modelul de transmitere ereditară a unui caracter monogenic în familie depinde de doi factori:
localizarea genei pe un autosom (caracter autosomal) sau pe cromosomii sexuali, X sau Y (caracter legat de X sau de Y);
fenotipul dominant sau recesiv determinat de genă.
Astfel există cinci modele de bază pentru transmiterea monogenică: autosomal dominant (AD), autosomal recesiv (AR), recesiv-legat de X (RLX), dominant-legat de X (DLX) şi legat de Y (LY). Anamneza familială poate indica şi alte modele de trnsnmitere, nemendeliene (de exemplu transmiterea maternală în bolile mitocondriale), dar la acestea ne vom referi distinct, ulterior.
Înainte de a descrie caracteristicile modelelor de transmitere monogenică sunt utile câteva consideraţii.
(1). Distincţia dintre ereditatea autosomală şi legată de X sau Y este evidentă dar trebuie subliniat că bărbaţii XY au un singur cromosom X şi deci nu sunt niciodată homozigoţi sau heterozigoţi pentru genele localizate pe cromosomii sexuali, ci numai hemizigoţi. De asemenea, între genele situate pe cei doi X, la femeile XX, şi genele de pe o pereche de autosomi apar diferenţe, deoarece ambele alele autosomale sunt active (exceptând fenomenul de amprentare, la care ne vom referi mai târziu) dar genele de pe X, deşi se găsesc în „doză dublă”, în marea lor majoritate sunt exprimate numai de pe unul din cei doi cromosomi X, celălalt fiind inactivat.
(2). Deşi există caractere determinate de gene/loci situate pe Y ele sunt foarte probabil neesenţiale pentru sănătate, deoarece femeile XX sunt perfect normale; aceste gene sunt implicate, în marea lor majoritate[35], numai în funcţiile sexuale, specific masculine, iar mutaţiile lor afectează numai aceste funcţii. Deci foarte probabil (exceptând bolile corelate cu funcţia sexuală masculină) la om nu există alte boli legate de Y
(3). Termenii de dominant şi recesiv sunt proprietăţi ale caracterelor şi nu ale genelor; ei se referă mai ales la fenotipul clinic, deşi descriptiv, sintetic, se folosesc termenii de alelă dominantă sau recesivă[36]. Clasic, distincţia dintre dominant şi recesiv se realizează la heterozigoţi: caracterele dominante se manifestă la heterozigoţi iar cele recesive nu se manifestă la heterozigoţi, ci numai la homozigoţi (sau heterozigoţi compuşi). Evident, la bărbaţii hemizigoţi pentru loci de pe X sau Y nu se pune problema de dominant sau recesiv. Diferenţa dintre ereditatea dominantă şi recesivă nu este absolută ci relativă (arbitrară) deoarece în multe boli recesive heterozigoţii au manifestări ale ambelor gene la nivel molecular/biochimic, celular şi uneori (în anumite condiţii) şi la nivel clinic. Să ne reamintim că sicklemia (vezi capitolul 4.B.1) este o afecţiune recesivă deoarece numai homozigoţii (a/a) manifestă obişnuit boala; heterozigoţii (N/a) prezintă însă trăsătura sicklemică deoarece produc atât HbA cât şi HbS, iar în anumite condiţii (scăderea concentraţiei de oxigen) pot manifesta şi semne clinice. Mai mult, mutaţii diferite în aceeaşi genă autosomală (de exemplu, gena pentru hormonul de creştere) se pot transmite ereditar diferit, unele dominant altele recesiv.
Deficienţa familială izolată de hormon de creştere (IGHD) – manifestată prin hipostatură severă – este produsă prin mutaţii diferite în gena hormonului de creştere (GH1) situată pe cromosomul 17. Formele recesive de IGHD sunt produse de mutaţii nonsens ce alterează structura produsului astfel că el nu poate fi secretat. La heterozigoţi, gena normală produce o cantitate de hormon de creştere redusă la jumătate dar suficientă pentru o talie cvasinormală; deficienţa se va manifesta numai la homozigoţi. În formele dominante de IGHD, la heterozigoţi, în alela mutantă se produce frecvent o mutaţie a situsului de clivare a intronului doi, care va deleta exonul trei al genei GH1; rezultă o proteină anormală, care se secretă (!); ea se combină (punţi disulfidice) cu proteina normală (codificată de alela normală), anulându-i efectele (dominanţă negativă) şi producând boala.
De aceea, în catalogul M.I.M, după anul 1994, caracterele autosomale au încetat să fie clasificate în dominante şi recesive.
În aceste condiţii se pune firesc întrebarea: cum vom diferenţia caracterele şi genele dominante? Distincţia este relativ simplă. Heterozigoţii cu o alelă normală şi alta mutantă vor avea doar jumătate din produsul normal al genei; dacă această jumătate este suficientă pentru realizarea funcţiilor genei, atunci alela mutantă este numită recesivă. Dacă acest lucru nu se întâmplă şi o alelă mutantă produce boala chiar dacă există o alelă normală, atunci alela mutantă este dominantă.
(4). Clasic, orice fenotip exprimat la heterozigoţi este definit ca dominant; această definiţie este prea rigidă deoarece în multe boli dominante homozigoţii (deşi foarte rari) sunt mai sever afectaţi decât heterozigoţii; pentru aceste cazuri, în care heterozigoţii au un fenotip intermediar între homozigoţii afectaţi şi cei normali, s-ar putea folosi termenii de dominaţă incompletă sau semidominanţă iar dacă ambele alele diferite se exprimă (de exemplu, în cazul alelelor HLA sau genelor A şi B pentru grupul sanguin ABO), termenul de codominanţă. Totuşi, în cazul bolilor monogenice aceşti termeni sunt rar folosiţi.
(5). Distincţia dintre dominant şi recesiv în cazul caracterelor/bolilor legate de X se estompează datorită fenomenului de inactivare (lyonizare) a unui cromosom X (inactivare precoce în viaţa embrionară, întâmplătoare şi definitivă). Într-adevăr, purtătoarele (heterozigotele Xa/XN) pentru boli recesive legate de X manifestă adesea unele semne, atunci când sunt comparate cu bărbaţii XY afectaţi, iar heterozigotele pentru boli dominante legate de X sunt mai uşor şi variabil afectate. Explicaţia este deja cunoscută (vezi capitolele 2.D.2.b şi 4.B.4): o femeie heterozigotă pentru un caracter legat de X este un mozaic de celule care exprimă fie alela normală, fie alela anormală; în funcţie de proporţia şi localizarea celulelor cu Xa activ apar semne variate de boală.
(6). În sfârşit, vom sublinia faptul că, în practica medicală, modelul de transmitere ereditară a unei boli poate fi rareori definit fără ambiguitate într-o singură familie! Familiile umane au, deobicei, un număr limitat de membri, mai ales când apar şi copii afectaţi, iar multe afecţiuni genetice pot fi consecinţa unei mutaţii noi sau pot prezenta fenomenul de heterogenitate genetică. De aceea, determinarea modului de transmitere, deşi este foarte importantă pentru sfatul genetic, este cel mai adesea o ipoteză de lucru şi nu un fapt stabilit. Numai atunci când gena a fost clonată şi mutaţia cauzatoare de boală poate fi determinată prin analiza ADN este posibilă stabilirea certă a modului de transmitere a mutaţiei. În celelalte cazuri un diagnostic precis ne permite să consultăm OMIM, unde intrările cu un mod de transmitere ereditară bine stabilit sunt notate cu asterisc.
(7) Pentru înţelegerea termenilor ce vor fi folosiţi în analiza transmiterii monogenice este necesară cunoaşterea datelor esenţiale privind structura şi funcţia genei, prezentate în capitolele 3.A şi 4.A, precum şi a terminologiei de bază (locus, alelă; homozigot, heterozigot simplu, dublu şi compus; dominant, codominant şi recesiv; pleiotropie, penetranţă, expresivitate; heterogenitate genetică etc, pe care cititorul îi poate găsi definiţi şi în glosarul de la sfârşitul cărţii)
2.2 TRANSMITEREA AUTOSOMAL DOMINANTĂ
Mai mult de 50% din intrările incluse în OMIM reprezintă caractere transmise autsomal dominant (AD) iar bolile AD afectează, la diverse vârste, circa 20 din 1000 de nou născuţi. Multe dintre ele sunt frecvente şi serioase prin consecinţele lor: hipercolesterolemia familială, boala polichistică renală-AD a adultului (ADPKD), neurofibromatoza 1, boala Huntington, polipoza adenoamtoasă familială, cancere ereditare de sân sau colon etc.
a). Caracteristici şi criterii de transmitere AD
În această situaţie, caracterul este determinat de o pereche de gene alele, situată în loci omologi, pe unul dintre autosomi. Una din genele alele suferă o mutaţie (A) şi se manifestă fenotipic (clinic) la heterozigoţi (A/n), deoarece alela normală (n) este insuficientă pentru a compensa efectul alelei mutante. Cele două alele A şi n pot forma trei genotipuri: A/n (mai frecvent), A/A (mai rar) şi n/n; primele două genotipuri vor determina un fenotip anormal (o boală) autosomal dominantă, în care barbaţii şi femeile vor fi egal afectaţi.
Teoretic, cu cele trei genotipuri se pot realiza şase combinaţii sau tipuri de încrucişări (tabel 5.6), dar practic, frecvenţa lor într-o populaţie depinde de severitatea manifestării clinice, care determină sau nu supravieţuirea până la vârsta adultă şi capacitatea reproductivă (fertilitatea). Evident primele două combinaţii sunt cele mai frecvente, a treia este rară şi, împreună cu următoarele trei (rarisime), se întâlnesc în familiile mari, numai pentru boli „uşoare” (de exemplu, brahidactilia, hemeralopia, prognatismul) sau pentru cele cu debut tardiv (după 40 de ani).
Tabel 5.6. Transmiterea dominantă autosomală a unui caracter patologic.
Sănătoşi (n/n) şi bolnavi (A/A; A/n)
|
Combinaţii genotipice parentale |
Gameţi |
Descendenţi |
|
|
Genotipuri |
Fenotipuri (indiferent de sex) |
||
|
1. n/n + n/n |
n x n |
n/n |
toţi sănătoşi |
|
2. A/n + n/n |
(A+n) x n |
½ A/n ½ n/n |
½ bolnavi ½ sănătoşi |
|
3. A/n + A/n |
(A+n) x (A+n) |
¼ A/A; ½ A/n ¼ n/n |
¾ bolnavi ¼ sănătoşi (indiferent de sex) |
|
4. A/A + n/n |
A x n |
A/n |
toţi bolnavi |
|
5. A/A + A/n |
A x (A+n) |
½ A/A; ½ A/n |
toţi bolnavi |
|
6. A/A + A/A |
A x A |
A/A |
toţi bolnavi |
Din analiza acestor combinaţii / încrucişări rezultă o serie de caracteristici (reguli) ale transmiterii autosomal dominante.
Doi indivizi sănătoşi (n/n + n/n) nu pot avea copii cu afecţiuni AD; boala nu este transmisă prin persoane neafectate deoarece, teoretic, nu există purtători sănătoşi de genă mutantă. Această regulă are o excepţie: producerea unor mutaţii (AD) germinale noi (în gametogeneza unuia dintre părinţii sănătoşi) poate determina apariţia unui copil afectat; există de asemenea „abateri” determinate de heterogenitatea genetică de locus (mutaţii diferite, dominate şi recesive, pot da acelaşi fenotip; de exemplu în retinita pigmentară; vezi caseta 3.6) sau de genotipul unuia dintre părinţii sănătoşi care este în realitate A/n dar alela A nu se exprimă deloc fenotipic (non-penetranţă) sau se va exprima mai târziu decât la copilul afectat (anticipaţie). În acelaşi context se pot include şi cazurile de non-paternitate (în care tatăl nelegitim este A/n).
Din încrucişarea dintre un bolnav heterozigot (A/n) şi un sănătos (n/n) rezultă, teoretic, 50% bolnavi (A/n) şi 50% sănătoşi (n/n):
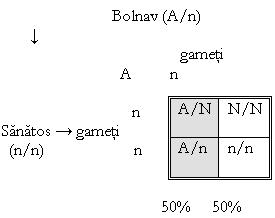
Rezultatul acestei încrucişări ne permite să formulăm alte reguli de bază ale transmiterii AD: fiecare bolnav are (cel puţin) un părinte afectat; cu alte cuvinte, boala este prezentă în fiecare generaţie şi în arborele genalogic al unei familii (suficient de mare) se observă o frecvenţă relativ ridicată a bolnavilor şi o transmitere verticală (figura 5.23). Aceste caracteristici, şi mai ales prezenţa, continuitatea, bolii în succesiunea generaţiilor, se observă frecvent în bolile AD mai putin severe, compatibile cu reproducerea şi, deseori, la cele cu debut tardiv.
Boala se manifestă (cu o frecvenţă şi o severitate relativ egală) atât la bărbaţi cât şi la femei iar gena mutantă (situată pe un autosom) poate fi moştenită şi transmisă cu o probabilitate egală de către ambele sexe (deci independentă de sex). Toate acestea pledează pentru o transmitere autosomală. În plus, transmiterea tată → fiu este posibilă; deşi acest criteriu nu este obligatoriu pentru transmiterea AD, prezenţa lui exclude alte tipuri de transmitere.
Evident, de la aceste reguli există şi excepţii, care complică interpretarea, şi pe care le vom discuta mai jos; este vorba de mutaţii noi, mozaicuri germinale, penetranţă redusă, anticipaţie, s.a.
Acum însă vom sublinia alt element important: fiecare bolnav, heterozigot (situaţia cea mai frecventă în bolile AD), are un risc de 50% de a avea descendenţi afectaţi, dacă gena are o penetranţă completă. Acest risc de 50% este valabil la fiecare sarcină (ca eveniment independent), indiferent dacă anterior s-a născut un copil normal sau afectat. Dar transmiterea gameţilor, la fel ca şi aruncarea unei monede, este supusă fluctuaţiilor întâmplării şi este posibil, spre exemplu, ca toţi copii (III-6, III-7, III-8, în figura 5.23) sau nici-unul dintre ei să fie afectaţi.
În transmiterea bolilor AD se poate întâlni uneori situaţia încrucişării dintre doi bolnavi heterozigoţi A/n x A/n (combinaţia 3 în tabelul 5.6) din care pot rezulta indivizi afectaţi şi homozigoţi (situaţiile 5 şi 6 din tabelul 5.6 sunt aproape numai teoretice). În multe boli AD (hipercolesterolemia familială, acondroplazia, sindromul Marfan, ş.a.) homozigoţii sunt mai grav afectaţi decât heterozigoţii şi deseori prezintă forme letale, neonatal, de boală. Spre deosebire de aceste boli cu semidominanţă (A/A > A/n > n/n) există şi cazuri de boli cu dominanţă completă (în care A/A = A/n) iar exemplul cel mai cunoscut fiind cel al bolii Huntington. Revenind la situaţia rară a încrucişării dintre doi bolnavi heterozigoţi vom semnala, ca o caracteristică a bolilor dominante, faptul că ei pot avea descendenţi săntoşi care, în bolile AD, vor fi şi fete (!) şi băieţi.
Pe baza datelor discutate mai sus se pot formula următoarele criterii privind transmiterea AD:
fiecare bolnav are de obicei un părinte afectat
orice bolnav transmite boala la jumătate din copiii săi; riscul de 50% este valabil la fiecare sarcină;
fenotipul anormal apare în fiecare generaţie (continuitate în succesiunea generaţiilor) iar în arborele genealogic se observă o transmitere verticală;
indivizii sănătoşi nu transmit boala;
Excepţiile de la aceste reguli sunt determinate de: penetranţa redusă, mutaţii noi sau mozaicuri germinale, debut tardiv de manifestare, non-paternitate şi, mai rar, heterogenitate genetică şi fenocopii;
boala se manifestă (cu o frecvenţă şi o severitate relativ egală) atât la bărbaţi cât şi la femei iar gena mutantă poate fi moştenită şi transmisă cu o probabilitate egală de către ambele sexe (deci independentă de sex; excepţia o constituie bolile care predomină sau sunt limitate la un sex).
transmiterea tată → fiu este posibilă şi caracteristică eredităţii AD.
doi bolnavi (cu o afecţiune compatibilă cu supravieţuirea şi reproducerea) pot avea descendenţi sănătoşi, fete şi băieţi, iar dintre copii afectaţi o parte pot fi homozigoţi, cu o formă mai gravă de boală.
Pe lângă aceste criterii majore ale transmiterii AD, în arborii genealogici există şi alte indicii pentru acest mod de transmitere: frecvenţa crescută a bolnavilor sau absenţa legăturilor consanguine.
b). Excepţii de la regulile transmiterii AD.
(1). Penetranţa redusă. În bolile AD, mai ales cele cu pleiotropie şi expresivitate clinică variabilă (vezi capitolul 4. A), uneori un individ moşteneşte alela mutantă dar nu o manifestă fenotipic; el poate însă să o transmită la descendenţi, care pot să fie afectaţi (figura 5.24.a; la subiectul II.1 gena mutantă este non-penetrantă, nu se manifestă clinic dar se transmite). Se realizează astfel un „salt” peste generaţia parentală, aparent sănătoasă. Dacă o genă mutantă se manifestă la toţi heterozigoţii (A/n) atunci are o penetranţă completă; dacă manifestarea ei este sub 100% atunci gena mutantă are o penetranţă redusă sau incompletă.
Penetranţa se poate măsura cantitativ prin determinarea proporţiei heterozigoţilor (A/n) care exprimă fenotipic gena mutantă din totalul „purtătorilor obligatorii” de mutaţie. În exemplul din figura 5.24.A penetranţa este de ¾ deci 75% sau 0,75. Cunoaşterea penetranţei este importantă în sfatul genetic deoarece riscul unui bolnav cu o afecţiune AD cu penetranţă redusă (de exemplu, 0,75) de a avea copii afectaţi este mai mic de 0,5 (50%), fiind 0,5 x 0,75 = 0.37. În acest context, vom preciza, din nou, că este foarte important să facem diferenţa dintre o mutaţie cu penetranţă redusă (cu risc suficient de mare, chiar dacă este sub cel teoretic de 50%) şi o mutaţie nouă (cu risc mic de recurenţă); în acest scop vom folosi toate mijloacele posibile de diagnostic presimptomatic.
Penetranţa este o noţiune cantitativă (de tipul „tot sau nimic”), care se referă la expresia clinică a genei mutante (prezentă sau absentă), ce poate fi influenţată de diferiţi factori: metoda de diagnostic, vârsta de debut, sexul.
Metoda de diagnostic este decisivă pentru a stabili dacă o genă se manifestă sau nu. De exemplu, în boala polichistică renală a adultului (ADPKD) o persoană tânără, ce a moştenit gena mutantă, poate să nu prezinte ecografic chisti renali vizibili (cu dimensiuni prea mici) dar ei ar putea fi puşi în evidenţă prin CT sau RMN. Într-un anumit sens, penetranţa poate fi considerată „un artefact” determinat de abilitatea noastră de a recunoaşte (clinic şi/sau paraclinic) expresia sau fenotipul genei mutante.
Vârsta de debut a primelor manifestări clinice este variabilă în multe boli AD (vezi mai jos); Vom menţiona aici că în boala Huntington penetranţa genei mutante este zero la naştere, 25% la 30 de ani, 50% la la 40 de ani şi 100% la 70 de ani.
Sexul persoanei poate unori influenţa manifestarea genei mutante; de exemplu, în cancerul de sân familial produs prin mutaţia genei BRCA2, penetranţa genei este redusă la bărbaţi al căror ţesut glandular este redus şi nedependent de stimularea hormonală.
Cauzele penetranţei reduse, incomplete, nu sunt clare dar intervin probabil factori genetici şi negenetici, care pot avea – într-o anumită conjunctură – un efect antagonic, „supresor”, asupra manifestării unei gene. Un exemplu edificator este deficienţa în factorul V Leiden, prezentată în caseta 5.5; vom mai adăuga faptul că în hipercolesterolemia familială depozitele de colesterol din pereţii vaselor sanguine, ce determină simptomatologia principală a bolii, pot fi reduse de o dietă sau o medicaţie adecvată (statine); vom aminti de asemenea „bolile induse de medicamente” (farmacogenetica) – ca porfiria acută intermitentă sau hipertermia malignă – care vor fi „declanşate” numai în anumite condiţii de mediu (vezi capitolul 8.A.3).
CASETA 5.5 OMI 227400.
DEFICIENŢA ÎN FACTORUL V LEIDEN
Un exemplu interesant şi util de ştiut îl furnizează deficienţa în factorul V Leiden, transmisă AD (gena este localizată pe cromosomul 1q23). Factorul V este o componentă a cascadei de coagulare a sângelui. Mutaţia Leiden (numită astfel după oraşul din Olanda în care a fost descoperită) produce o alterare a funcţiei factorului V. Heterozigoţii pentru alela factorului V Leiden prezintă o stare de hipercoagulabilitate, care determină un risc crescut de producerea unor tromboze venoase profunde (TVP) la membrele inferioare[37], de unde cheagurile se pot desprinde şi ajunge, prin circulaţie, în plămâni producând o embolie pulmonară (EP). Fenotipul clinic TVP se manifestă numai la circa 50% din heterozigoţi, deci nu orice persoană care moşteneşte gena mutantă va dezvolta boala deoarece trebuie să intervină alţi factori de risc pentru TVP şi EP: folosirea de contraceptive orale (!), sedentarismul, sarcina, prezenţa unui cateter venos central, alţi factori de mediu sau genetici ce contribuie la hipercoagulabilitatea sângelui; toate acestea explică penetranţa redusă a genei.
(2). Mutaţii germinale noi. Pacienţii afectaţi pot fi primul şi singurul caz de boală AD din familie; în această situaţie se presupune că apariţia bolnavului din părinţi sănătoşi şi homozigoţi (n/n) (figura 5.24.c) este consecinţa unei mutaţii spontane noi (neomutaţii) apărută în timpul gametogenezei, de obicei la bărbat; această ipoteză se va lua în consideraţie după ce s-au eliminat celelalte posibilităţi: penetranţa redusă, expresivitatea variabilă şi non-paternitatea.
De altfel, mutaţiile noi sunt frecvente în numeroase boli AD care, datorită gravităţii handicapului clinic sau reducerii până la anulare a capacităţii reproductive, nu pot fi transmise de la părinţi la descendenţi. În osteogenesis imperfecta tipul II (OMIM 166200), boală AD – caracterizată prin fragilitate osoasă – letală perinatal, toate cazurile reprezintă mutaţii noi. Un alt exemplu frecvent citat este al acondroplaziei (OMIM 100800) – un nanism disproporţionat pe seama membrelor, foarte scurte – în care se apreciază că 80% din cazuri sunt produse prin neomutaţii. În neurofibromatoză acestea reprezintă 50% din cazuri.
(3). Mozaicismul germinal. Mult mai rar, mutaţiile se pot produce precoce în viaţa embrionară, afectând unii din precursorii gameţilor; se formează astfel o sub-populaţie de gonocite ce poartă mutaţia, deci un mozaicism germinal care poate explica apariţia mai multor copii afectaţi din părinţi sănătoşi (figura 5.24.d). Considerat foarte rar, acest mecanism începe să fie probat prin analizele moleculare: de exemplu, a fost demonstrat în 6% din cazurile de copii cu osteogenesis imperfecta născuţi din părinţi sănătoşi, precum şi în alte boli (neurofibromatoză sau unele boli recesive-legate de X).
c). Expresivitatea variabilă.
Expresivitatea, spre deosebire de penetranţă, este o noţiune calitativă, care defineşte gradul, intensitatea, natura, severitatea manifestării fenotipice a unei gene mutante şi penetrante la bolnavi diferiţi. Atunci când aceşti parametri sunt diferiţi pentru expresia unei gene mutante se foloseşte termenul de expresivitate variabilă (figura 5.24.b).
Expresivitatea variabilă interesează:
spectrul de manifestarea a simptomelor şi distribuţia lor,
vârsta de debut a semnelor,
intensitatea (severitatea) exprimării,
limitarea manifestării la pacienţii de un anumit sex.
(1). Spectrul de manifestare al simptomelor şi distribuţia lor. Numeroase boli AD, mai ales cele cu pleiotropie (manifestări variate în organe multiple) prezintă o expresivitate variabilă. În capitolul 4.A.4.2 exemplificam acest concept la bolnavii cu sindrom Marfan (caseta 4.1), la care pot să nu apară toate anomaliile celor trei sisteme afectate (schelet, ochi, aparat cardiovascular) sau la care severitatea clinică este diferită; prezentăm acum un arbore genealogic ilustrativ (figura 5.25), în care toţi indivizii vii afectaţi au aceiaşi mutaţie în gena fibrilinei dar prezintă o variabilitate marcată în vârsta de debut, distribuţia şi severitatea semnelor. Un alt exemplu ilustrativ este cel al neurofibromatozei (NF1) sau boala Von Recklinghausen (vezi capitolul 11.E.1) care prezintă manifestări cutanate (multiple pete café au lait, pistrui axilari, ş.a), oftalmologice (noduli Lisch în iris, ş.a), musculoscheletice (scolioză, statură mică ş.a.), neuropsihiatrice (dificultăţi de învăţare sau retard mintal, convulsii etc) şi tumorale (neurofibroame de diverse mărimi, glioame de căi optice sau SNC, feocromocitoame ş.a). Toate aceste modificări se dezvoltă la vârste diferite (primele semne ce apar la copii sunt petele multiple café au lait) şi au o mare variabilitate a distribuţiei severităţii.
Modul şi gradul (severitatea) de manifestare a unei boli AD determinată de o anumită genă mutantă nu poate fi prezis, nici chiar la bolnavii din aceeaşi familie, care au aceeaşi mutaţie genică. În schimb, cunoaşterea întregului spectru de manifestare al unei boli monogenice AD permite evaluarea şi diagnosticul complet al unui pacient, precum şi cercetarea unor cazuri oligo- sau mono-simptomatice, puţin evidente, în familie. Toate acestea vor influenţa calitatea diagnosticului, conduita medicală, prevenţia unor complicaţii şi acurateţea sfatului genetic.
(2). Vârsta de debut a semnelor. O problemă corelată cu expresivitatea variabilă este vârsta de debut a manifestărilor unor boli AD pleiotrope. Diversele semne apar în timp, la vârste diferite (penetranţă dependentă de vârstă); uneori primele manifestări ale unor boli (ca ADPKD, hipercolesterolemia familială, polipoza de colon, boala Huntington ş.a) apar tardiv la adult. În figura 5.26 prezentăm exemplul clasic al coreei Huntington – o boală degenerativă neurologică caracterizată prin mişcări anormale (coree) şi pierderea progresivă a funcţiilor mintale (demenţă) – în care primele semne apar, în medie la 35 de ani. Persoana care a moştenit gena mutantă este mult timp asimptomatică, fără să ştie că posedă mutaţia în momentul în care îşi alcătuieşte o familie şi decide să aibe copii (cărora, evident, le poate transmite gena anormală). Acest lucru scoate în evidenţă utilitatea anamnezei familiale şi importanţa unor teste de diagnostic presimptomatic (vezi capitolul 18).
În acest context, vom menţiona şi fenomenul de anticipaţie: în unele afecţiuni AD semnele de boală se manifestă în generaţii succesive la o vârstă tot mai precoce şi cu o severitate tot mai mare. Mult timp existenţa anticipaţiei a fost discutabilă dar astăzi se ştie că este un fenomen cert, mai ales în bolile prin amplificarea repetiţiilor trinucleotidice (vezi capitolele 6.B.1.4 şi 11.C), în care numărul acestor expansiuni instabile va creşte în succesiunea generaţiilor determinând o vârstă de debut tot mai precoce şi o severitate mai mare.
(3). Limitarea manifestării la pacienţii de un anumit sex. Expresivitatea variabilă a unor boli poate să se manifeste şi prin afectarea predominantă (sau chiar limitată) a unui anumit sex. Ne refeream mai sus la cancerul de sân familial produs prin mutaţia genei BRCA2 csre este foarte rar la bărbaţi, dar sunt şi alte exemple de boli AD manifeste la un anumit sex; vom mai cita: porfiria acută intermitentă predominantă la sexul feminin sau pubertatea precoce familială limitată la sexul masculin.
În acest ultim exemplu, pubertatea apare precoce la 4-5 ani, datorită unei mutaţii a receptorului pentru hormonul luteinizant care, aşa cum se observă în figura 5.27, se transmite AD (vezi transmiterea tată → fiu) dar gena este nemanifestă la persoanele de sex feminin (I-1, II-1,II-5) care o moştenesc şi o transmit la descendenţi.
Cauzele expresivităţii variabile sunt greu de definit. Sigur, la bolanvii cu aceiaşi afecţiune dar din familii diferite, prima explicaţie a expresivităţii variabile poate fi heterogenitatea alelică, deci existenţa unor mutaţii diferite. De exemplu, în osteogenesis imperfecta mutaţiile care afectează aminoacizii dinspre capătul carboxil al moleculei de procolagen vor da forme mai grave de boală decât cele care afectează aminoacizii dinspre capătul amino terminal.
Pentru bolnavii din aceeaşi familie sau din familii diferite care au aceeaşi mutaţie cauzele fenomenului sunt, însă, altele. Se invocă interacţiunea genei-boală cu alte gene modificatoare (caseta 5.6) şi/sau cu unii factori de mediu; o dovadă indirectă a existenţei genelor modificatoare o reprezintă expresia mai frecventă/predomi nantă sau exclusivă/limitată a unor gene autosomale la un anumit sex.
2.3 TRANSMITEREA AUTOSOMAL RECESIVĂ
Circa 1/3 din caracterele monogenice sunt transmise autosomal recesiv (AR). Cele mai frecvente boli AR în populaţia europaeană sunt hemocromatoza şi fibroza chistică sau mucoviscidoza. Alte afecţiuni AR: surditatea congenitală (unele forme), fenilcetonuria, atrofia musculară spinală, cecitatea recesivă, sindromul adrenogenital, sicklemia, talasemia, albinismul oculo-cutanat, etc. Marea majoritate a erorilor înăscute de metabolism se trasnmit de asemnea AR:
CASETA 5.6.
Genele modificatoare
Multe boli mendeliene prezintă diferenţe în ceea ce priveşte vârsta de apariţie, severitatea bolii şi alte caracteristici fenotipice. De fapt, expresia unitară/identică a unor asemenea boli pare sa fie excepţia şi nu regula (o asemenea exceptie pare sa fie de exemplu albinismul, boală genetică determinată de deficienţa completă a tirozinazei). Printre factorii care determină această diversitate fenotipică sunt implicate variantele alelice, mediul şi genele modificatoare. Exemplele privind variantele alelice şi factorii de mediu sunt relativ numeroase şi bine caracterizate. Existenţa unor gene modificatoare a fost iniţial postulată o data cu descrierea unor diferenţe fenotipice la purtătorii aceleiaşi variante alelice şi în absenţa intervenţiei unor factori de mediu. Asemenea gene modificatoare pot afecta penetranţa, expresivitatea şi pleiotropia sau pot modifica dominanţa unor mutaţii. In funcţie de natura efectului lor, genele modificatoare pot determina fenotipuri mai severe, mai puţin severe sau chiar fenotipuri noi. Epistazia, care apare atunci când o alelă a unei gene maschează fenotipul unei alte gene, este o formă de modificare genetică. Deşi sunt probabil extrem de numeroase, numărul genelor modificatoare bine documentate la om este în prezent relativ redus.
Gene modificatoare ale penetranţei. Deşi penetranţa incompletă este o trăsătură comună a unor numeroase boli mendeliene, există puţine exemple pentru care să se ştie că variabilitatea pentranţei se datorează unor gene independente. Un exemplu a fost descris în cazul surdităţii non-sindromice determinată de mutaţii ale genei DFNB26 de pe cromosomul 4q31. Au fost descrise cazuri în care indivizi homozigoţi pentru mutaţia prezentă în familie au totuşi auz normal. La aceşti indivizi a fost cartografiată o gena modificatoare dominantă localizată pe cromosomul 7 care reduce penetranţa mutaţiei cauzatoare de boală. De asemeni, pe cromosomul 8, în apropierea markerului D8S277, a fost localizată o genă care modifică penetranta mutaţiei 1555AG de la nivelul genei mitocondriale pentru ARNr 12S, ce produce surditatea transmisă matriliniar.
Gene modificatoare ale dominanţei. Una dintre mutaţiile implicate în producerea retinitei pigmemtare cu transmitere autosomal recesivă este cea a genei pentru periferina-1 (PRP1). Au fost însă descrise cazuri în care mutaţii heterozigote ale acestei gene se însoţesc de prezenţa bolii, deşi se aşteapta ca numai indivizii homozigoţi să fie bolnavi. Aceste cazuri au fost explicate prin asocierea unor variante alelice la nivelul genei neînlanţuite ROM1 (care codifică proteina 1 a porţiunii externe a celulelor cu bastonaş de la nivelul retinei). Ca urmare, o alelă care are în mod normal un efect recesiv în producerea retinitei pigmentare poate sa acţioneze ca o alelă dominantă atunci când se asociază o alela particulară a genei neînlanţuite ROM1.
Expresivitatea variabilă. Un exemplu de genă modificatoare implicată în producerea expresivităţii variabile este cel al hipercolesterolemiei familiale, boală transmisă autosomal dominant care afectează una din 500 de persoane din populatia generală şi determină o creştere semnificativă a riscului pentru bolile cardiovasculare ca urmare a creşterii colesterolemiei. S-a constatat că unii purtători heterozigoţi pentru mutaţia cauzatoare de boală au un nivel al colesterolemiei care este cu 25% mai mic decât cel aşteptat. La asemenea indivizi a fost cartografiată o genă modificatoare localizată pe cromosomul 13q.
Pleiotropia. Febra mediteraneeana familială (FMF) este o boală autosomal recesivă comună în regiunea mediteraneeană caracterizată prin febră recurentă şi inflamaţia seroaselor. Boala este determinată de mutaţii ale genei pentru pirina/marenostrina. O complicaţie variabilă a acestei boli este asocierea amiloidozei renale ce conduce la insuficienţă renală. Unele studii au aratat ca asocierea variabilă a acestei complicaţii este explicată, cel puţin în parte, de unele variante polimorfice la nivelul genei pentru amiloidul A seric. De asemeni, o genă modificatoare a apariţiei ileusului meconial în cadrul fibrozei chistice a fost localizată la nivelul cromosomului 19q13.
Deşi pe de o parte fenomene precum dominanţa incompletă, expresivitatea variabilă sau pleiotropia complică analiza genetică a caracterelor mendeliene, identificarea genelor modificatoare responsabile de asemenea fenomene se poate dovedi sursa unor noi metode de diagnostic, tratament şi prevenţie a bolilor genetice.
a). Caracteristici şi criterii de transmitere AR
O genă recesivă îşi exercită efectul numai în stare homozigotă (a/a), întrucât în stare heterozigotă (N/a) boala nu apare deoarece efectul alelei normale (N) este suficient pentru realizarea funcţiei, cu toate că alela mutantă (a) produce o reducere a funcţiei controlată de locusul respectiv. Pentru a se realiza starea homozigotă individul bolnav primeşte o genă mutantă de la fiecare din părinţii săi. Astfel, în cazul afecţiunilor recesive, spre deosebire de cele dominante, moştenirea se face prin ambii părinţi care, în cea mai mare parte a cazurilor, sunt sănătoşi şi purtători de genă mutantă, deci heterozigoţi: N/a + N/a. După cum se observă din tabelul 5.7 (poziţia 1) în descendenţa lor rezultă teoretic un raport fenotipic de 3 sănătoşi la 1 bolnav; acest raport se verifică însă numai în cazul unor afecţiuni mai frecvente, puţin severe, şi în familiile mari sau cumulând datele obţinute de la mai multe familii[38].
Tabel 5.7. Transmiterea recesivă autosomală a unui caracter patologic.
Sănătoşi (N/N ; N/a) şi bolnavi (a/a)
|
Combinaţii genotipice parentale |
Gameţi |
Descendenţi |
|
|
Genotipuri |
Fenotipuri |
||
|
1. N/a x N/a |
(N + a) x (N + a) |
¼ N/N ; ½ N/a ¼ a/a |
¾ sănătoşi din care 2/3 purtători ¼ bolnavi, indiferent de sex. |
|
2. N/a x N/N |
(N + a) x (N) |
½ N/N ; ½ N/a |
Toţi sănătoşi, ½ purtători |
|
3. a/a x N/N |
(a) x (N) |
N/a |
Toţi sănătoşi şi purtători |
|
4. a/a x N/a |
(a) x (N + a) |
½ N/a; ½ a/a ; |
½ sănătoşi şi purtători, ½ bolnavi |
|
5. a/a x a/a |
(a) x (a) |
a/a |
Toţi bolnavi |
Din datele prezentate mai sus se poate formula o regulă importantă a transmiterii autosomal recesive (AR): doi părinţi sănătoşi, heterozigoţi pentru aceeaşi pereche de gene alele, pot avea copii (fete şi băieţi) bolnavi cu o anomalie AR. De subliniat faptul că ambele sexe sunt afectate în proporţii cvasi-egale şi pot transmite alela mutantă cu aceiaşi probabilitate (elemente logice pentru localizarea autosomală a genei).
Deoarece părinţii bolnavului sunt sănătoşi, în arborele genealogic (figura 5.28) se înregistrează o discontinuitate a bolii în succesiunea generaţiilor. Bolnavul poate fi singurul membru afectat al familiei[39]; dacă apar şi alte cazuri, ele se găsesc frecvent în aceeaşi fratrie, dând aspectul unei transmiteri orizontale.
Riscul unui cuplu de indivizi normali şi heterozigoţi de a avea un copil bolnav (a/a) este de 25% sau ¼ la fiecare sarcină, ca eveniment independent. Ar fi greşit să credem că în familiile cu patru copii este obligatoriu ca
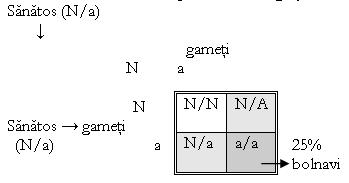
numai unul să fie afectat, sau mai rău, dacă s-au născut trei copii sănătoşi al patrulea va fi bolnav. Riscul de ¼ este valabil la fiecare sarcină; să ne imaginăm o loterie cu patru bile, trei albe şi una neagră, din care la fiecare naştere s-ar extrage o bilă; înaintea naşterii următoare „bila se pune la loc” şi tragerea se repetă. Uneori pot exista familii cu şansa de a avea 1-4 copii normali, alteori se pot naşte succesiv doi bolnavi.
În afara combinaţiei a doi heterozigoţi, alte încrucişări din care pot rezulta copii bolnavi sunt trecute în tabelul 5.7, în poziţiile 4 şi 5. Ocazional, un bolnav (a/a) se poate încrucişa cu un sănătos heterozigot (N/a) şi atunci 50% din descendenţi ar putea fi afectaţi; pe arborele genealogic se înregistrează un apect de pseudo-dominanţă, boala fiind prezentă în două generaţii (figura 5.29). În sfârşit, mult mai rar, din încrucişarea dintre doi bolnavi, homozigoţi pentru aceeaşi mutaţie (a/a x a/a) vor rezulta numai descendenţi bolnavi (a/a). Acest tip de uniune „asortativă” se observă în colectivităţi speciale (deficienţi vizuali, surzi ş.a) şi pentru boli ce nu afectează capacitatea reproductivă. De la regula enunţată mai sus fac excepţie situaţiile de heterogenitate genetică în care bolnavii cu afecţiuni asemănătoare clinic au mutaţii în gene (loci) diferite (figura 5.30); întrucât părinţii bolnavi nu sunt homozigoţi pentru aceeaşi mutaţie descendenţii lor vor fi dublu heterozigoţi şi sănătoşi.
În afecţiunile autosomal recesive se observă că printre părinţii bolnavului există o mare proporţie mai mare de căsătorii între persoane care au cel puţin un strămoş comun (consanguinitate), în special între veri primari.
În concluzie, criteriile majore pentru diagnosticul eredităţii recesive autosomale la om sunt următoarele:
marea majoritate a persoanelor afectate sunt urmaşi ai părinţilor aparent sănătoşi dar heterozigoţi; riscul lor de a avea un copil bolnav este 1/4, indiferent de sex;
dacă bolnavii se căsătoresc cu persoane normale şi homozigote toţi copiii vor fi sănătoşi; se înregistrează astfel aspectul de discontinuitate a bolii în succesiunea generaţiilor sau de cazuri izolate;
dacă un bolnav se căsătoreşte cu o persoană sănătoasă şi heterozigotă, riscul de a avea descendenţi bolnavi, fete şi băieţi, este de 50% (pseudo-dominanţă); în acest caz, transmiterea mamă-fiică este posibilă.
doi părinţi bolnavi (homozigoţi pentru aceleaşi gene) nu vor avea copii sănătoşi.
părinţii copilului afectat sunt mai frecvent rude, comparativ cu părinţii copiilor sănătoşi (deci frecvenţa crescută a consanguinităţii).
În afara criteriilor enunţate mai sus, bolile AR se caracterizează prin faptul că sunt, în general, anomalii prin lipsă (hipoplazie, hipofuncţie, deficit enzimatic etc), deseori mai grave decât cele AD.
b). Consanguinitatea în anomaliile recesive.
Probabilitatea ca două persoane să fie purtătoare (N/a) ale aceleiaşi mutaţii creşte dacă persoanele respective sunt înrudite pentru că fiecare poate moşteni gena mutantă de la un strămoş comun; această situaţie este numită consanguinitate şi se notează în arborele genealogic printr-o linie dublă.
S-a observat că frecvenţa consnguinităţii parentale în bolile AR este invers proporţională cu frecvenţa genei recesive: cu cât boala este mai rară, cu atât consanguinitatea la părinţi este mai frecventă. Astfel în cazul unei afecţiuni cu o frecvenţă de 1:10.000 (albinismul) circa 6% dintre părinţii copiilor bolnavi vor fi veri primari, faţă de 1% cât există în populaţia generală europeană. Procentul va creşte la 16% dacă afecţiunea este de 10 ori mai rară (1:100.000).
Părinţii înrudiţi posedă un procentaj mai mare de gene identice, comparativ cu populaţia generală. Acest procent depinde de gradul de înrudire, fiind de 1/8 la verii primari, 1/32 la verii de gradul II ş.a.m.d (tabelul 5.8)
Tabelul 5.8 Coeficientul de înrudire (r) şi consanguinitate (F)
în diferite tipuri de încrucişări între rude.
|
Tip de încrucişare |
Coeficient de înrudire (r) |
Coeficient de consanguinitate (F) |
|
Tată –fiică; frate – soră; gemeni dizigoţi |
1/2 |
1/4 |
|
Unchi – nepoată; demi-frate – demi-soră |
1/4 |
1/8 |
|
Veri de gradul I; demi-unchi – demi-nepoată |
1/8 |
1/16 |
|
Văr de gradul I – vară de gradul II; demi-veri primari |
1/16 |
1/32 |
|
Veri de gradul II |
1/32 |
1/64 |
|
Văr gradul II – vară gradul III |
1/64 |
1/128 |
|
Veri de gradul III |
1/128 |
1/256 |
Coeficientul de înrudire (r) este proporţia de gene comune (identice) din totalul genelor persoanelor ce au un strămoş comun. Coeficientul de consanguinitate (F) este probabilitatea ca un urmaş să fie homozigot pentru gene ce provin de la părinţi, cu un anumit grad de rudenie, de la un strămoş comun. F = r/2. Cei doi coeficienţi se calculează prin metode speciale.
Deoarece părinţii înrudiţi au un număr mai mare de gene (normale sau anormale) în comun, consanguinitatea creşte riscul de întâlnire a heterozigoţilor şi favorizează apariţia subiecţilor homozigoţi afectaţi. Astfel, dacă într-o populaţie frecvenţa heterozigoţilor pentru o afecţiune recesivă este 1/50, probabilitatea unei persoane de a avea un copil bolnav căsătorindu-se cu o persoană neînrudită este 1/50 x 1/50 x 1/4 = 1/10.000; dacă s-ar căsători cu vara sa primară atunci riscul naşterii unui copil afectat este 1/50 x 1/8 x 1/4 = 1/1.600, deci de circa şase ori mai mare.
Se apreciază că riscul global al unui descendent anormal din căsătoria între veri primari este totuşi mic (3-5%), aproape dublu faţă de cel al populaţiei generale (2-3%). Consecinţele unui incest (încrucişarea dintre rude de gradul I) sunt însă mult mai dezastruoase: ½ – ¼ din descendenţi vor fi anormali (retardul mintal fiind pe locul întâi).
c). Heterozigoţii în anomaliile recesive.
Unul dintre cele mai importante aspecte legate de transmiterea AR îl constituie studiul şi depistarea heterozigoţilor (N/a). Ei sunt fenotipic sănătoşi şi atunci când se căsătoresc cu un alt heterozigot sănătos au un risc de 25% de a avea un copil bolnav. Problema devine „acută” în familiile în care există un individ afectat de o boală recesivă. Probabilitatea ca o persoană să moştenescă în acest caz gena autosomală depinde de gradul înrudirii cu bolnavul (figura 5.31): cu cât este mai apropiat, cu atât riscul este mai mare. Astfel fratele/sora unui bolnav cu o afecţiune recesivă are o probabilitate de 2/3 de a fi heterozigot; aceasta se reduce la 1/3 pentru copilul lor sănătos (nepot al bolnavului)
Întrucât genele recesive se transmit nemanifeste din generaţie în generaţie, până ce se realizează starea homozigotă (ca şi râurile subterane care atunci când afluează ţâşnesc la suprafaţă), există riscul ca oricine să posede astfel de gene şi deci să fie heterozigot. Acest risc se poate estima pe baza frecvenţei afecţiunii în populaţie (vezi capitolul 7) şi se poate calcula aproximativ după formula:
h = 2 √ frecvenţa boli; ![]()
deci frecvenţa heterozigoţilor este de două ori rădăcina pătrată a frecvenţei afecţiunii în populaţie. De exemplu, dacă fenilcetonuria are o frecvenţă de 1:10.000, frecvenţa heterozigoţilor este de 1/50. Prevalenţa heterozigoţilor pentru unele afecţiuni AR mai frecvente este de 1/10 pentru hemocromatoză (1:400); 1/25 pentru fibroza chistică sau mucoviscidoză (1:2.500); 1/30 pentru surdimutitate (1:3.600); 1/35 pentru sindromul adreno-genital (1:5.000). Se observă că în populaţie numărul heterozigoţilor este foarte mare comparativ cu cel al bolnavilor homozigoţi recesivi. În aceste condiţii posibilitatea întâlnirii a doi heterozigoţi este mare; astfel, se estimează că în Europa de Vest 1:625 cupluri este alcătuit din heterozigoţi pentru fibroza chistică. Genele recesive morbide sunt deci relativ comune şi, pentru că sunt multe afecţiuni recesive este probabil că fiecare dintre noi să purtăm câteva gene anormale în stare heterozigotă.
În anumite populaţii frecvenţa bolilor recesive şi deci cu atât mai mult a heterozigoţilor este foarte mare: sicklemia în Africa, beta-talasemia în Italia şi Grecia, hemocromatoza şi fibroza chistică/mucoviscidoza în Europa de Vest şi Nord, nefroza congenitală în Finlanda, boala Tay-Sachs la evreii Ashkenazi (originari din centrul şi estul Europei). Explicaţiile acestui fenomen (avantajul selectiv al heterozigoţilor, efectul de fondator, izolarea etnică sau religioasă etc) vor fi discutate în capitolul 7.
Aşa cum am mai spus, noţiunile de dominant şi recesiv sunt valabile pentru fenotip, pentru caracter, şi reprezintă o simplificare a relaţiilor moleculare, deoarece ambele alele ale genelor se manifestă întotdeauna la nivel molecular. Acest lucru permite depistarea heterozigoţilor, sănătoşi dar purtători, pentru acele afecţiuni la care se cunoaşte efectul primar al genei şi la care acesta poate fi evidenţiat. Ne vom referi pe larg la acest aspect important al profilaxiei bolilor genetice (în capitolul 18.4) dar acum vom reaminti exemplul sicklemiei (capitolul 4.B.1) în care heterozigoţii sunt identificaţi, simplu, prin electroforeza hemoglobinei, care evidenţiază HbA şi HbS.
d). Heterozigoţii compuşi.
Heterogenitatea alelică este un fenomen frecvent în bolile AR: o genă poate suferi mutaţii alelice diferite. De exemplu, în beta-talasemie s-au identificat numeroase mutaţii în gena HBB; deseori la bolnavi cele două alele ale locilor HBB sunt anormale (a/a) dar au mutaţii diferite (a1/a2) şi aceştia sunt de fapt heterozigoţi compuşi (a1/a2). Se pare că în cele mai multe din bolile recesive indivizii afectaţi sunt în fapt mai curând heterozigoţi compuşi decât homozigoţi adevăraţi, exceptând evident situaţiile în care părinţii lor sunt înrudiţi şi au moştenit aceeaşi mutaţie de la un strămoş comun sau când în acea populaţie predomină o anumită mutaţie, ca urmare a efectului de fondator.
e). Variabilitatea în bolile recesive.
Bolile recesive prezintă deseori o penetranţă completă şi o variabilitate clinică redusă. Există însă şi excepţii: în hemocromatoza (vezi caseta 3.1) de pildă nu toţi bolnavii (a/a) dezvoltă boala iar femeile sunt relativ protejate datorită pierderilr de fier din cursul menstruaţiei. De asemenea în mucoviscidoză bolnavii pot avea o mare variabilitate a distribuţiei şi severităţii manifestărilor digestive, pulmonare etc (vezi capitolul 11.E.2), explicabilă prin mutaţii diferite, specifice, ale genei CFTR; homozigoţii care au mutaţia F508del prezintă insuficienţă pancreatică dar în alte mutaţii funcţia pancreatică este cvasi-normală, fenomenele pulmonare reduse sau se observă o absenţă congenitală a canalelor deferente.
Mutaţiile noi sunt rare în bolile AR şi, de regulă, se poate presupune că ambii părinţi ai unui copil afectat sunt heterozigoţi. Totuşi 1% din cazurile de atrofie musculară spinală (SMA) tip I (boala Werdnig-Hoffman) copilul afectat moşteneşte o alelă de la unul din părinţi care este heterozigot iar cealaltă alelă provine printr-o mutaţie germinală nouă de la un părinte ne-purtător; în aceste cazuri riscul de recurenţă este foarte mic.
Aşa cum am precizat mai sus heterogenitatea alelică este frecventă în bolile AR; dar şi heterogenitatea de locus se întâlneşte in unele afecţiuni AR. Multe din surdităţile congenitale sau formele de cecitate recesivă sunt produse de mutaţii în loci autosomali diferiţi.
Vom menţiona, în sfârşit, fenomenul de disomie uniparentală prin care un copil afectat poate moşteni ambele copii ale unui cromosom (cu alela mutantă) de la unul din părinţi, dar asupra acestui fenomen, rar dar interesant, vom reveni mai târziu.
2.4 TRANSMITEREA LEGATĂ DE SEX
Transmiterea legată de sex se referă la ereditatea genelor situate pe cromosomii sexuali. După cum ştim deja, cromosomii X şi Y au morfologie, gene, roluri şi distribuţie diferită la cele două sexe. Prima diferenţă majoră este reprezentată de dimensiunea şi numărul redus de gene al cromososomului Y (aproape 2/3 din Yq este reprezentat de ADN înalt repetitiv, inactiv genetic). Marea majoritate a genelor sunt implicate în determinismul sau controlul unor caractere sexuale, specific masculine, şi nu au alele omologe pe X. Numai o mică parte din genele cromosomului Y, situate mai ales în regiunile pseudo-autosomale, au alele omologe[40] pe X şi pot realiza recombinări în cursul meiozei. Cromosomul X (cu peste 1100 de gene) are gene de sexualizare dar şi gene nesexuale. O a doua diferenţă majoră este reprezentată de faptul că genele de pe X se vor găsi la organismele feminine (XX) în doză dublă (homozigote sau heterozigote) în timp ce la bărbaţi (XY) vor fi în doză unică (hemizigot). Această diferenţă de dozaj este compensată (aproape complet) prin inactivarea unuia din cei doi cromosomi X de la femeie.
Ţinând cont de datele precizate sintetic mai sus – este logic să presupunem că există mai multe modele particulare de transmitere legată de sex:
transmiterea caracterelor legate de Y;
transmiterea caracterelor determinate de genele regiunilor pseudo-autosomale;
transmiterea cararacterelor legate de X, recesivă sau dominantă.
a). Transmiterea legată de Y (holandrică)
Un caracter legat de Y se transmite de la tată, prin cromosomulY, la toţi fii săi. Se cunosc extrem de puţine caractere patologice cu acest mod de transmitere (de exemplu, o formă de retinită pigmentară care se transmite de la tată exclusiv la băieţi; OMIM 400004). Mutaţiile genelor implicate în determinismul şi diferenţierea sexuală (dintre care cel mai bine cunoscute sunt mutaţiile genei SRY ) produc o inversiune sexuală şi/sau sterilitate. În schimb alelele polimorfice ale ADN din cromosomul Y se transmit după modul holandric precizat mai sus.
b). Transmiterea caracterelor determinate de gene din regiunile pseudo-autosomale X şi Y.
Pentru a recunoaşte un caracter determinat de o genă din această regiune, trebuie studiată ascendenţa şi descendenţa subiecţilor de sex masculin. Cei care au moştenit caracterul de la tatăl lor, prin intermediul cromosomului Y, îl vor transmite mai frecvent la fii lor, în timp ce cei care l-au primit de la mama lor; prin intermediul cromosomului X, îl vor transmite preferenţial fetelor lor. Acest mod de ereditate a fost numită ereditate parţial legată de sex.
Până în prezent se cunoaşte un singur exemplu: discondrosteoza Leri-Weil, o displazie scheletică cu statură mică disproporţionată şi deformarea antebraţului, produsă de mutaţia genei SHOX, genă pseudoautosomală care nu este inactivată, ce codifică un factor de transcripţie implicat în reglarea creşterii. În arborele genealogic (figura 5.32) frecvenţa crescută a bolii la femei sugerează o transmitere dominantă legată de X dar prezenţa unei transmiteri tată (II3) → fiu (III-5) exclude o ereditate legată strict de X.
c).Transmiterea legată de X
Pe cromosomul X au fost localizate peste 1100 de gene din care circa 50% se asociază cu fenotipuri anormale. Ele au o distribuţie diferită la cele două sexe şi un mod de transmitere particular, uşor de identificat. Luând în discuţie o genă mutantă situată pe X sunt posibile cinci genotipuri: XNY şi XaY – la bărbat, XNXN, XNXa şi XaXa – la femeie. Mutaţia genei se exprimă la bărbat (hemizigot) indiferent dacă manifestarea ei fenotipică este dominantă sau recesivă. La femeile heterozigote XNXa, datorită fenomenului de inactivare întâmplătoare a cromosomului X, alela mutantă se exprimă în 50% din celule indiferent dacă mutaţia produce un fenotip recesiv (cel mai frecvent) sau dominant (rar); deci distincţia fenotipică între transmiterea dominantă şi recesivă se estompează (prin „hemizigoţie funcţională) şi este arbitrară, condiţionată de proporţia femeilor heterozigote care manifestă boala şi severitatea ei. În plus, unele mutaţii legate de X se pot asocia cu inactivarea preferenţială a cromosomului Xa la femei, fenomen care poate atenua consecinţele fenotipice. De aceea mulţi geneticieni nu mai clasifică bolile legate de X în recesive legate de X (RLX) şi dominante legate de X (DLX). Considerăm însă că această împărţire este utilă deoarece fenotipurile dominante se exprimă constant la heterozigote în timp ce, în mod obişnuit, fenotipurile recesive nu se manifestă.
2.5. Transmiterea recesivă legată de X
Dintre bolile RLX cele mai cunoscute sunt hemofilia, distrofia musculară Duchenne, cecitatea pentru culori, deficienţa în G6PD ş.a.
O mutaţie RLX se manifestă fenotipic la bărbaţii hemizigoţi XaY (frecvent) şi la femeile homozigote XaXa (foarte rar); femeile sănătoase pot fi XNXN sau XNXa (purtătoare de genă mutantă sau conductoare). Cu cele cinci genotipuri sunt posibile următoarele combinaţii (tabelul 5.9) dar în funcţie de boală, în practică, se întâlnesc numai o parte dintre acestea:
Tabel 5.9. Transmiterea recesivă legată de X a unui caracter patologic.
Sănătoşi (XNY, XNXN , XNXa) şi bolnavi (XaY, XaXa)
|
Combinaţii genotipice parentale |
Gameţi |
Descendenţi |
|
|
Genotipuri |
Fenotipuri |
||
|
1. XNY x XaXN |
(XN + Y) x (Xa + XN) |
¼ XaY ; ¼ XNY, ¼ XaXN ;¼ XNXN |
Un sfert din copii vor fi bolnavi (aceştia sunt ½ băieţi), toate fetele vor fi sănătoase (½ purtătoare) |
|
2. XaY x XNXN |
(Xa + Y) x (XN + XN) |
XNY ; XaXN |
Toţi copii vor fi sănătoşi dar toate fetele vor fi purtătoare |
|
3. XaY x XaXN |
(Xa + Y) x (Xa + XN) |
¼ XaY ; ¼ XNY, ¼ XaXa ; ¼ XaXN, |
½ băieţi sunt bolnavi ½ fete sunt bolnave, ½ fete sunt purtătoare |
|
4. XNY x XaXa |
(XN + Y) x (Xa + Xa) |
XaY ; XaXN |
Toţi băieţii bolnavi, toate fetele purtătoare |
|
5. XaY x XaXa |
(Xa + Y) x (Xa + Xa) |
XaY ; XaXa |
Toţi copiii (băieţi şi fete) bolnavi |
|
6. XNY x XNXN |
(XN + Y) x (XN + XN) |
XNY ; XNXN |
Toţi copiii sănătoşi |
a). Criteriile transmiterii recesive legate de X.În bolile RLX, de obicei, bolnavi sunt numai bărbaţii, care au primit gena mutantă de la femei sănătoase dar purtătoare (combinaţia 1, tabel 5.9). Deci, din părinţi sănătoşi rezultă copii bolnavi (1/4) , dar aceştia vor fi numai băieţi (1/2), care au primit gena anormală de la mamă; fetele vor fi sănătoase (½ purtătoare). În arborele genealogic (figura 5.33) se înregistrează o transmitere oblică, în diagonală, şi discontinuă, de la un bărbat bolnav (bunic, străbunic), prin femei sănătoase dar purtătoare, la un băiat bolnav.
Dacă bolnavii au capacitate de reproducere (de exemplu, în hemofilie) şi se căsătoresc cu o femeie sănătoasă şi homozigotă (combinţia 2) nu pot transmite boala băieţilor (transmiterea tată → fiu nu este deci posibilă) sau prin băieţi la generaţiile următoare. În schimb toate fetele vor fi obligatoriu heterozigote, purtătoare.
În cazul unor mutaţii frecvente şi puţin severe ale unor gene de pe X, cum ar fi deficienţa în G6PD (în populaţia Afro-Americană 10% din bărbaţi au deficienţă de G6PD iar 18% din femei sunt heterozigote) sau daltonismul (8% din bărbaţi şi 1:150 din femei) este posibil ca bolnavii (XaY) să se căsătorească cu o femeie heterozigotă (combinaţia 3, tabel 5.9)[41]; în descendenţa acestui cuplu pot apărea băieţi bolnavi (1/2) dar şi fete bolnave (1/2). În arborele genealogic (figura 5.34) se observă transmiterea tată → fiu dar alela mutantă a băieţilor provine…de la mamă. În aceleaşi afecţiuni sunt uneori posibile şi combinaţiile 4 şi 5 din tabelul 5.9. Se observă fapul că o femeie bolnavă va transmite boala la toţi băieţii (situaţie caracteristică transmiterii RLX) iar din doi părinţi bolnavi rezultă numai copii bolnavi (situaţie sugestivă pentru transmiterea recesivă, în general).
În concluzie, criteriile majore pentru transmiterea RLX sunt următoarele:
bolnavii sunt aproape exclusiv bărbaţi; ei nu transmit niciodată boala;
femeile heterozigote sunt deobicei neafectate şi pot avea băieţi bolnavi, indiferent de tipul de căsătorie.
gena mutantă se transmite de la bărbaţi afectaţi → la femei sănătoase şi purtătoare → şi de la acestea la ½ băieţi; deci există o discontinuitate în succesiunea generaţiilor, doi părinţi sănătoşi (mama heterozigotă) putând avea copii bolnavi, dar numai băieţi
nu este posibilă niciodată transmiterea tată → fiu (deoarece tatăl îi dă fiului cromosomulY). (când se observă o astfel de situaţie va trebui să ne gândim la o boală AD limitată la sexul masculin; de exemplu, scleroza glomerulară focală, din figura 5.35)
b). Mutaţii noi
Multe boli RLX sunt severe şi nu permit supravieţuirea până la vârsta reproducerii. Distrofia musculară Duchenne (DMD) este exemplul cel mai elocvent. Alelele mutante se pierd astfel din populaţie odată cu bolnavul; şi totuşi prevalenţa bolii nu se modifică deoarece alte cazuri apar prin mutaţii noi, din femei sănătoase şi cert nepurtătoare. În DMD circa 1/3 din cazuri apar din cupluri de părinţi sănătoşi, în care femeia nu este heterozigotă; în hemofilie selecţia este doar parţială si numărul de mutaţii noi este mult mai mic.
c). Femei afectate de boli recesive legate de X
Bolile recesive legate de X se manifestă aproape exclusiv la sexul masculin. Prezenţa certă a unei astfel de boli la o persoană de sex feminin are multiple explicaţii.
Încrucişarea între un bolnav şi o femeie purtătoare (combinaţia 3, tabel 5.9)(figura 5.33); aşa cum am spus această eventualitate, rară, se întâlneşte mai ales în bolile RLX frecvente în anumite populaţii şi care sunt mai puţin severe.
Inactivarea „neechilibrată” a cromosomului X la femeile heterozigote Xa/XN, care face ca o proporţie mai mare de celule să aibă activ Xa.
Inactivarea „preferenţială” a unui cromosom X anormal structural (isocromosom X sau translocaţie X-autosom); acest tip de inactivare reflectă mai probail o selecţie în favoarea celulelor care au XN activ şi deci nu există în realitate.
Femei hemizigote pentru X datorită unei monosomii 45,X (sindromul Turner).
Femei 46,XY în cadrul unor stări intersexuale (vezi capitolul 14.C).
Mutaţie nouă (pe cromosomul XN) la femei purtătoare Xa/XN.
Heterogenitate genetică.
d). Riscul genetic. Depistarea heterozigotelor pentru gene RLX
Riscul de recurenţă al bolilor recesive legate de X depinde de genotipul părinţilor şi sexul descendenţilor. Se poate calcula din tabelul 5.7 dar problema esenţială este de a şti dacă o femeie sănătoasă din familia/fratria unui bolnav este sau nu purtătoare. Acest lucru (frecvent în practică pentru distrofia musculară Duchenne) se poate face, pornind de la anamneza familială, pe baza faptului că femeile heterozigote sunt, datorită inactivării unuia din cromosomii X, un mozaic funcţional, cu celule în care este activ XN şi celule în care este activ Xa şi efectul primar sau secundar al genei mutante se poate identifica la nivel biochimic sau celular. O altă modalitate este analiza directă sau indirectă la nivelul ADN.
2.6. Transmiterea dominată legată de X.
Bolile DLX sunt mult mai puţine decât cele RLX şi, cu excepţia particulară a sindromului X fragil, ele sunt foarte rare. Exemplul cel mai cunoscut este rahitismul hipofosfatemic (rezistent la vitamina D) în care se produce o eliminare crescută a fosfaţilor în urină (datorită unei tulburări de reabsorbţie renală), statură mică şi deformări osoase. Alte afecţiuni DLX sunt: condrodisplazia punctata, incontinentia pigmenti, sindromul Goltz, sindromul Rett (despre care se pot obţine detalii prin accesarea OMIM)
Aşa cum am precizat, o boală legată de X este considerată dominantă dacă se exprimă regulat la femeile heterozigote (XAXn). La bărbaţi nu se pune problema diferenţierii între un caracter dominant sau recesiv legat de X deoarece ei sunt hemizigoţi. Bărbaţii bolnavi (XAY) vor prezenta însă forme mai grave de boală deoarece la femeile afectate, în marea lor majoritate heterozigote (XAXn), inactivarea întâmplătoare face ca 50% din celule să aibe cromosomul Xn activ, fapt ce determină un fenotip mai puţin sever. În unele boli (de exemplu, incontinentia pigmenti[42] tipul 2, sindromul Rett) manifestarea este aproape exclusivă la sexul feminin, deoarece boala este letală la feţii XAY.
Tabel 5.10. Transmiterea dominantă legată de X a unui caracter patologic.
Sănătoşi (XnY, XnXn ) şi bolnavi (XAY, XAXn, XAXA)
|
Combinaţii genotipice parentale |
Gameţi |
Descendenţi |
|
|
Genotipuri |
Fenotipuri |
||
|
1. XnY x XAXn |
(Xn + Y) x (XA + Xn) |
XAXn, XnXn XAY, XnY |
½ fete bonave şi ½ fete sănătoase ½ băieţi bolnavi şi ½ sănătoşi |
|
2. XAY x XnXn |
(XA + Y) x (Xn) |
XAXn, XnY |
Toate fetele bolnave, Toţi băieţii sănătoşi |
|
3. XAY x XAXn |
(XA + Y) x (XA + Xn) |
XAXA, XAXn XAY, XnY |
Toate fetele şi ½ băieţi bolnavi 1/2 băieţi sănătoşi |
|
4. XnY x XAXA |
(Xn + Y) x (XA) |
XAXn, XAY |
Toţi copiii (băieţi şi fete) bolnavi |
|
5. XAY x XAXA |
(XA + Y) x (XA) |
XAXA, XAY |
Toţi copiii (băieţi şi fete) bolnavi |
|
6. XnY x XnXn |
(Xn + Y) x (Xn) |
XNY ; XNXN |
Toţi copiii sănătoşi |
Modelul de transmitere în bolile DLX nu diferă esenţial de transmiterea AD deoarece femeile au o pereche de cromosomi X ca ori ce altă pereche de autosomi; apar, evident, o serie de particularităţi determinate de localizarea genei pe cromosomul X (tabelul 5.10)(figura 5.36). Vom regăsi deci continuitatea bolii în succesiunea generaţiilor, determinată de faptul că orice bolnav are (cel puţin) un părinte bolnav. Fiecare copil al unei femei afectate şi heterozigote (XAXn) are un risc de 50% de a fi bolnav, indiferent de sex (combinaţia 1 din tabelul 5.10). Părinţii sănătoşi vor avea copii sănătoşi (combinaţia 6). Ceea ce este însă caracteristic transmiterii DLX este faptul că bărbaţii bolnavi (XAY) vor avea toate fetele afectate şi toţi băieţii sănătoşi (combinaţia 2; transmiterea tată → fiu este imposibilă) iar în situaţia, mai mult teoretică, a doi bolnavi (femeia heterozigotă) (combinaţia 3) pot rezulta copii sănătoşi, dar numai băieţi. Aceste caracteristici determină o frecvenţă generală a femeilor bolnave de două ori mai mare ca a bărbaţilor bolnavi.
3. ereditatea monogenicĂ nON – mendelianĂ
Transmiterea caracterelor monogenice, prin modelele sale şi raporturile de segregare ale genelor alele este, în general, concordantă cu principiile eredităţii mendeliene. Au fost, totuşi, semnalate o serie de excepţii care sunt determinate de mecanisme neobişnuite: mutaţii instabile, disomie uniparentală, amprentare genomică, mozaicism sau ereditate mitocondrială. Ele sunt discutate în detalii şi în alte capitole, dar pentru o viziune unitară a problemei vom face şi aici unele referiri, redundanţa fiind, în opinia noastră, benefică.
3.1. MUTAŢIILE INSTABILE sau DINAMICE
Se consideră, în general, că alelele mutante ce produc boli monogenice mendeliene se transmit nemodificate de la părinţi la copii. În anul 1991 au fost descoperite mutaţiile prin expansiunea unor repetiţii trinucleotidice instabile (vezi capitolul 6.B.1.4) care reprezintă un mecanism genetic nou pentru un grup important de boli (vezi capitolul 11.D).
Au fost identificate mai multe gene ce conţin regiuni cu repetiţii trinucleotidice (de exemplu, CGG în sindromul X fragil, CTG în distrofia miotonică sau CAG în boala Huntington. În populaţia generală, numărul de repetiţii variază de la o persoană la alta; când numărul lor depăşeşte o anumită limită, regiunea în care se găsesc devine instabilă şi are tendinţa să crească în mărime (premutaţie), atunci când este transmisă la descendenţi. Expansiunea neobişnuită a repetiţiilor transformă premutaţia în mutaţie completă şi produce apariţia manifestărilor clinice ale bolii. Deoarece vârsta de debut şi severitatea bolii sunt direct propoRţionale cu mărimea expansiunii, acest fenomen explică fenomenul de anticipaţie clinică observat în acest grup de boli, unde în aceiaşi familie vârsta debutului clinic scade în generaţii succesive. În unele afecţiuni, gravitatea bolii depinde de sexul părintelui ce transmite gena mutantă; astfel transmiterea maternă pentru distrofia miotonică şi sindromul X fragil sau paternă pentru boala Huntington – produc forme mai severe de boală.
3.2. DISOMIA UNIPARENTALĂ
Un alt mecanism neobişnuit implicat în producerea bolilor genetice este disomia uniparentală (DUP), în care ambele copii ale unui anumit cromosom provin numai de la unul din părinţi. Deobicei DUP se produce prin pierderea unui cromosom de către un zigot care a fost iniţial trisomic. În funcţie de originea trisomiei (nedisjuncţie în mieoza I sau în meioza II) (vezi figura 5.20)şi cromosomul pierdut există trei posibilităţi:
zigot disomic normal, cu câte un cromosom de la fiecare din părinţi;
zigot cu DUP şi doi cromosomi identici de la un părinte (isodisomie);
zigot cu DUP şi doi cromosomi diferiţi de la un părinte (heterodisomie).
DUP nu are consecinţe clinice prin ea însăşi ci prin faptul că isodisomia poate realiza o stare de homozigoţie a unei gene recesive ce există sub formă heterozigotă numai la unul din părinţi. S-au descris cazuri de fibroză chistică în care ambele gene mutante provin de la două copii identice ale cromosomului 7 matern. De asemenea, au fost semnalate cazuri de hemofilie A în care ambii cromosomi X şi Y ai unui băiat bolnav provin de la tatăl afectat sau boala se manifestă la o fată homozigotă (prin isodisomia Xa) născută din părinţi XaXN şi XNY.
DUP poate avea efecte patologice atunci când cromosomul implicat conţine una sau mai multe gene amprentate. De exemplu, în sindromul Beckwith-Wiedemann (OMIM 130650) – caracterizat prin gigantism, macroglosie, omfalocel şi predispoziţie pentru dezvoltarea unor tumori – boala este asociată cu expresia bialelică (prin isodisomie) a genei pentru factorul de crestere similar insulinei tip 2 (IGF2), genă situată pe cromosomul 11p15.5 şi exprimată în mod normal[43] numai de pe cromosomul patern (bolnavul are ambii cromosomi 11 de la tată).
3.3. AMPRENTAREA PARENTALĂ (GENOMICĂ).
În transmiterea mendeliană se pleacă de la premiza că acţiunea genei este independentă de originea sa maternă sau paternă; când se formează un zigot, genele “par a uita” originea lor parentală. În mod obişnuit o alelă mutantă a unei gene autosomale este transmisă cu o probabilitate egală de la unul din părinţi, indiferent de sex, la un copil de un sex sau altul. Există însă situaţii rare în care expresia unei gene mutante şi deci a bolii este diferită în funcţie de originea ei, maternă sau paternă. Această diferenţă de expresie între alela moştenită de la tată şi alela moştenită de la mamă este produsă prin amprentare parentală sau genomică (vezi capitolul 4.D.1.2). Amprentarea este o formă de inactivare a genei ce produce o expresie monoalelică dar nu este o mutaţie, deoarece este reversibilă.
Amprentarea se produce în gametogeneză, inainte de fecundare, marcând originea maternă sau paternă a a numitor gene. După concepţie expresia genei amprentate este suprimată în anumite ţesuturi şi se menţine ca atare toată viaţa; excepţie fac celulele germinale în care se produce o conversie a amprentării funcţie de sex: la bărbat, alelele moştenite de la mamă, cu amprentare maternă, vor fi convertite în gametogeneză şi vor fi trecute descendenţilor cu amprentare paternă.
Amprentarea este un mecanism normal de reglarea genică care nu produce, prin el însăşi, o boală ereditară dar poate explica penetranţa incompletă sau expresivitatea variabilă a unei afecţiuni monogenice.
De exemplu, tumora de glomus carotidian, transmisă AD, la care subiecţii purtători de genă mutantă sunt bolnavi sau sănătoşi după cum ei au primit gena de la tată sau de la mamă; deci gena mutantă este activă dacă o transmite tatăl sau inactivă dacă provine de la mamă.
Amprentarea genomică implică numai un număr mic de gene. Sunt cunoscute mai multe regiuni cromosomice (care conţin peste 40 de gene amprentate) situate pe: 7q matern, 11p patern, 14q matern, 15q matern, 15q patern, etc. În ciuda numărului relativ mic de gene amprentate, tulburarea modului corect de expresie monoalelică (datorată UPD, deleţiei cromosomice / genice sau pierderii amprentării) s-a dovedit a avea consecinţe serioase în dezvoltarea embrionară, boli genetice şi cancer. Cel mai amplu exemplu studiat (vezi capitolul 4.D.1.2) îl reprezintă sindroamele Prader-Willi (SPW) şi Angelman (SA) produse, în mod obişnuit (dar nu exclusiv ; vezi tabelul 5.11), de aceeaşi deleţie a cromosomului 15q11-13, dar pe cromosomul de origine paternă (SPW) sau maternă (SA).
Tabel 5.11. Fenotipurile şi cauzele sindroamelor Prader-Willi şi Angelman
|
|
Sindromul Prader – Willi |
Sindromul Angelman |
|
Fenotip |
Hipotonie neonatală severă, dismorfie facială caracteristică, mâini şi picioare mici, talie mică, obezitate cu debut în copilărie, retard mintal, tulburări de comportament |
Dismorfie facială carcteristică, talie mică, microcefalie, retard mintal sever, spasticitate şi convulsii, lipsa vorbirii şi crize de râs. |
|
Deleţie 15q11-13 |
~ 70% cazuri cu deleţie pe cromosomul patern |
~ 70% cazuri cu deleţie pe cromosomul matern |
|
Disomie uniparentală |
~ 30% cazuri (maternă) |
~ 3-5 % cazuri (paternă) |
|
Gene incriminate |
SNRPN şi necdina, ambele cu expresie de pe cromosomul patern. |
UBE3A care este exprimată exclusiv matern, la nivel cerebral. |
|
Mutaţie genică |
Neidentificată |
7-9 % cazuri |
|
Mutaţie în centrul de amprentare |
1-2 % cazuri |
7-9 % cazuri |
|
Alte cauze |
Neidentificate |
10-20 % cazuri |
3.4. MOZAICISMUL
Mozaicismul este definit ca prezenţa la un individ a cel puţin două linii celulare diferite genetic dar derivate dintr-un singur zigot (prin aceasta se deosebeşte de himeră, formată prin unirea a doi zigoţi ce vor forma un singur embrion; vezi 5.C.3).
Mozaicismul prezintă următoarele caracteristici:
poate implica un cromosom întreg (aneuploidie) sau o singură genă (mutaţie);
este un eveniment postzigotic care se produce într-o singură celulă;
o dată ce modificarea genetică a fost generată şi nu este letală, ea va fi trasnsmisă celulelor fiice, formându-se o clonă celulară anormală;
procesul se poate produce în perioade diferite ale dezvoltării ontogenetice: în stadiul embrionar, în viaţa fetală sau postnatal; momentul de producere va determina proporţia relativă a celor două clone şi prin aceasta severitatea modificărilor fenotipice cauzată de clona anormală.
Nu vom discuta mozaicismul cromosomic, produs de o eroare postzigotică în mitoză (vezi 4.B.2.3) şi nici nu vom mai insista asupra mozaicismului funcţional care se întâlneşte la femei, prin inactivarea unuia din cei doi cromosmi X. Ne vom referi doar la mutaţiile genice produse în celulele somatice, care pot produce un fenotip segmental sau neuniform (în „pete”). Mozaicismul somatic este o cauză majoră a multor forme de cancer (vezi capitolul 17) dar a fost semnalat şi în unele boli monogenice „clasice”, ca de exemplu neurofibromatoza 1 segmentală (ce afectează numai o parte a corpului iar părinţii pacientului sunt normali) în unele boli legate de X (hemofilia A, distrofia musculară Duchenne, deficienţa în ornitin transcarbamilază) sau în unele tulburări de morfogeneză.
Există şi un mozaicism germinal sau gonadic care explică transmiterea unei boli genetice la mai mulţi copii de către părinţi normali. Mutaţia se produce în perioada precoce a dezvoltării embrionare (este deci o mutaţie somatică) în celulele precursoare ale gameţilor, care efectuează un număr de diviziuni mitotice (câteva sute la embrionul masculin), ceea ce face ca o anumită proporţie de gameţi să posede mutaţia. Mozaicismul germinal a fost documentat în circa 6% din formele letale de osteogenesis imperfecta (figura 5.37), la 15% din mamele (nepurtătoare !) copiilor cu distrofie musculară Duchenne sau în unele cazuri de hemofilie A. Posibilitatea mutaţiilor germinale face dificilă excluderea completă a riscului de recurenţă în boli recesive legate de X, la care testele de identificare a stării de purtător la mamă sunt normale, sau în bolile autosomal dominante în care părinţii sunt clinic şi genetic neafectaţi dar din fericire acest risc este mic (3-4%).
3.5. transmiterea pe linie maternĂ
În capitolul 2.C.3 am prezentat structura ADN mitocondrial şi am precizat că acesta poate suferi mutaţii producând o serie de boli degenerative, care sunt transmise într-un mod specific, de la mamă la toţi descendenţii; bărbaţii bolnavi nu transmit boala. Explicaţia acestui mod particular de transmitere, pe linie maternă, este reprezentată de faptul că numai ovulul va furniza zigotului citoplasmă şi mitocondrii. Bolile produse de mutaţii în ADNmt vor fi prezentate în capitolul 12.
E. EREDITATEA POLIGENICĂ ŞI MULTIFACTORIALĂ
Numeroase caractere normale şi anormale (calitative sau cantitative) sunt prezente la mai mulţi indivizi dintr-o familie, desi nu sunt determinate monogenic (tabelul 5.12). Această agregare familială poate fi explicată prin faptul că membrii aceleiaşi familii au un număr mai mare de gene în comun. Dar aceasta nu este singura explicaţie a unor trăsături comune într-o familie. Nu trebuie uitat că membrii unei familii trăiesc în aceleaşi condiţii de mediu socio-economic şi cultural şi nici posibilitatea ca, uneori, o aceeaşi boală să apară din întâmplare la mai multe persoane din aceiaşi familie. Totuşi, majoritatea caracterelor familiale non-mendeliene sunt determinate de acţiunea combinată a unor factori genetici, reprezentaţi de mai multe gene nealele (poligenie), şi a unor factori de mediu. Deci, ereditatea lor este complexă, multifactorială.
O caracteristică primară a bolilor multifactoriale este aceea că indivizii afectaţi tind să se grupeze în familii (agregare familială). Specialiştii în epidemiologie genetică trebuie să stabilească dacă o agregare familială observată în anumite boli este determinată de factori genetici, să evalueze contribuţia lor şi în final să identifice locii şi alelele implicate în aceste boli multifactoriale.
Tabelul 5.12. Caractere multifactoriale normale şi anormale
|
Caractere normale |
Caractere anormale |
|
culoarea pielii şi a părului culoarea ochilor înălţimea dermatoglifele coeficientul de inteligenţă habitusul fizionomia feţei personalitatea individului
|
– despicătura labio-palatină defectele de tub neural malformaţiile congenitale de cord luxaţia congenitală de şold schizofrenia boala coronariană diabetul zaharat – cancerul de sân, cancerul de colon, etc |
4.1. Stabilirea naturii genetice a unui caracter familial non-mendelian
Stabilirea naturii genetice a unui caracter familial non-mendelian, care ar putea fi un caracter multifactorial, se realizează prin trei metode esenţiale: determinarea şi măsurarea agregării familiale, studiul gemenilor şi studiile de adoptaţie.
a). Măsurarea agregării familiale
Cea mai directă metodă pentru evidenţierea unui determinism genetic este să stabilim că un anumit caracter calitativ/boală este familial, se manifestă la mai mulţi membri din familie. Când doi indivizi dintr-o familie au aceeaşi boală ei se numesc concordanţi pentru boala respectivă. Când, într-o pereche de indivizi înrudiţi, numai unul este afectat şi celălalt nu, ei se numesc discordanţi pentru boala dată. Gradul de agregare familială poate fi măsurat prin compararea frecvenţei caracterului / bolii la rudele unei persoane afectate (proband) cu frecvenţa în populaţia generală. Rata riscului relativ ( λr ) este definită prin raportul dintre prevalenţa bolii la rudele unei persoane afectate (r) şi prevalenţa bolii în populaţia generală.
|
λr = |
Prevalenţa bolii la rudele unei persoane afectate “r” |
|
Prevalenţa bolii în populaţia generală |
Valoarea lui λr exprimă nivelul agregării familiale în funcţie de riscul recurenţei bolii în familie şi de prevalenţa ei în populaţia generală[44]. Cu cât λr este mai mare cu atât agregarea familială este mai pronunţată (tabelul 5.13). O rată λr = 1 indică faptul că persoana înrudită nu are un risc mai mare decât orice individ din populaţia generală de a dezvolta boala. Rata riscului relativ se poate măsura pentru diferite clase de înrudire; de exemplu, pentru gemeni MZ, rude de gradul I (λf pentru fratrie) etc.
Tabelul 5.13.Rata de risc relativ ( λr ) pentru rudele probanzilor
cu unele boli familiale şi ereditate multifactorială.
(adaptat după Nussbaum et al, 2001)
|
Boala |
Gradul de înrudire |
λr |
|
Autism |
Gemeni monozigoţi Fraţi |
2000 150 |
|
Diabet zaharat tip I |
Gemeni monozigoţi Fraţi |
80 12 |
|
Psihoza maniaco-depresivă |
Gemeni monozigoţi Fraţi |
60 7 |
|
Schizofrenia |
Gemeni monozigoţi Fraţi |
48 12 |
Este firesc ca incidenţa unei boli multifactoriale şi λr să fie concordante cu gradul de rudenie (tabelul 5.14). Dacă există o predipoziţie genetică la o boală este firesc ca λr să fie mai mare la gemenii monozigoţi (MZ) – cu aceiaşi ereditate – şi apoi să scadă la rudele de gradul I (cu ½ gene în comun) şi, mai mult , la rudele de gradul II (cu ¼ gene în comun).
Tabelul 5.14. Riscul familial în schizofrenie
(modificat după Strachan şi Read, 1999)
|
Grad de rudenie |
Incidenţă |
λr |
|
Copii |
12,31 % |
15,4 |
|
Fratrie |
8,51 % |
10,6 |
|
Nepoţi, nepoate |
2,81 % |
3,5 |
|
Veri primari |
2,91 % |
3,6 |
|
Unchi, mătuşe |
2,01 % |
2,5 |
O altă metodă de evaluare a agregării familiale este reprezentată de studiile caz-control. În care pacienţii cu boală (cazurile) sunt comparaţi (istoric familial, expuneri la factori de mediu, ocupaţie, poziţie geografică, paritate etc) cu indivizi fără boală (control) (de exemplu soţiile, ce trăiesc în acelaşi mediu). Pentru a determina o posibilă contribuţie genetică la agregarea familială a bolii, se compară frecvenţa bolii în familia cazului cu frecvenţa ei la rudele lotului control. Studiile caz-control sunt dificile şi pasibile la erori de recenzare, alegerea loturilor etc.
În cazul caracterelor cantitative ele nu mai pot fi clasificate în „prezente sau absente” ci vor trebui măsurate. În consecinţă nu se mai poate compara prevalenţa bolii la rude versus control ci se măsoară coeficientul de corelaţie, adică tendinţa ca o anumită valoare a măsurătorii paramerului analizat să fie asemănătoare în loturile evaluate
b). Studiile gemenilor
Pentru evaluarea rolului eredităţii şi mediului la om, gemenii sunt subiecţi ideali, adevărate “experimente naturale” (Fr. Crick sugera, cu ironie, că fiecare pereche de gemeni ar trebui…donată ştiinţei pentru studiu). Gemenii dizigoţi (DZ) permit măsurarea concordanţei bolilor la doi indivizi înrudiţi care cresc şi trăiesc în acelaşi mediu dar care sunt diferiţi din punct de vedere genetic, proporţia de gene comune fiind de 50%. Gemenii monozigoţi (MZ) au genotip identic şi pot creşte şi trăi împreună sau nu. Dacă vor trăi în condiţii de mediu diferite acestea vor fi responsabile de diferenţele în apariţia bolilor în cursul vieţii lor.
Gemenii monozigoţi provin prin clivajul timpuriu în embriogeneză (în primele 14 zile după concepţie[45]) al unui singur zigot. Ei vor avea întotdeauna acelaşi sex şi vor fi identici din punct de vedere genetic exceptând posibilele mutaţii somatice post-zigotice (mai frecvente în dezvoltarea celulelor sistemului imun), numărul moleculelor de ADN mitocondrial sau modelul de inactivare a cromosomului X (dacă sunt de sex feminin). Gemenii dizigoţi se dezvoltă din două ovule diferite, fiecare dintre ele fertilizate de către un spermatozoid; au acelaşi sex sau sexe diferite (în proporţii egale) şi, din punct de vedere genetic, sunt diferiţi sau tot atât de asemănători ca fraţii obişnuiţi, având numai jumătate din alelele lor identice (se mai numesc şi gemeni fraterni). Frecvenţa naşterilor gemelare variază între 0,5 – 2 %. În general se apreciază că 1/3 din toţi gemenii sunt MZ iar 2/3 sunt DZ din care 1/2 sunt de acelaşi sex, 1/2 sunt de sex diferit.
Diagnosticul gemelarităţii, necesar pentru gemenii de acelaşi sex, necesită evaluarea atentă a multor caracteristici: aspectul placentei, numărul membranelor amniotice şi corionice; morfologia corpului, dermatoglifele; markerii serologici şi, cel mai sigur, markerii ADN polimorfici. Toţi gemenii dizigoţi au doi saci amniotici şi doi saci corionici (ei pot fuziona secundar dar circulaţia fiecărei părţi a placentei rămâne separată). Natura membranelor fetale la gemeniii MZ depinde de momentul separării. La 75% din MZ există un singur amnios şi un singur corion; restul de 25% au două membrane corionice şi nu pot fi deosebiţi în acest caz de gemenii DZ. Diagnosticul de gemelaritate este pus de baza unor markeri serologici şi ADN identici.
Studiile gemenilor sunt adesea folosite pentru a distinge trăsăturile multifactoriale de cele monogenice sau de cele cauzate de factori negenetici:
Deoarece gemenii monozigoţi au toate genele lor comune, ei vor fi 100% concordanţi pentru transmiterea tuturor bolilor monogenice. Gemenii dizigoţi vor avea o concordanţă de 50% pentru trăsăturile dominante monogenice şi de 25% pentru cele recesive.
O concordanţă sub 100% la gemenii MZ este o dovadă clară că factori negenetici au un rol în producerea bolii şi că avem de a face mai degrabă cu o trăsătură multifactorială.
Concordanţa trăsăturilor multifactoriale este diferită după tipul de gemelaritate. (Tabel 5.15). Cu cât procentul de concordanţă la MZ este mai mare decât la DZ cu atât o componentă genetică a bolii este mai pronunţată. Această observaţie este cu atât mai valabilă cu cât boala este manifestă la naştere sau cât mai curând după naştere. Cu cât debutul este mai tardiv, cu atât posibilitatea intervenţiei unor factori de mediu este mai mare.
Gemenii MZ despărţiţi la naştere şi crescuţi separat permit geneticienilor o bună observaţie a concordanţei îmbolnăvirilor la indivizi identici genetic, crescuţi sub influenţa unor factori de mediu diferiţi. Asemenea studii au permis demonstrarea rolului mediului în producerea unor boli psihice cum ar fi: alcoolismul sau abuzul de droguri.
Tabel 5.15 Concordanţa unor trăsături între gemenii monozigoţi şi dizigoţi
(Emery and Rimoin, 1997)
|
Caracter |
Procentul de concordanţă |
|
|
Gemeni monozigoţi |
Gemeni dizigoţi |
|
|
Diabet zaharat insulino-independent |
100 |
10 |
|
Înălţimea |
95 |
52 |
|
Nr. crestelor digitale |
95 |
49 |
|
IQ Psoriazis Epilepsia idiopatică |
90 72 70 |
60 15 6 |
|
Psihoza maniaco-depresivă |
67 |
5 |
|
Retard mintal cu IQ < 50 |
60 |
3 |
|
Schizofrenia |
53 |
15 |
|
Diabet zaharat insulino-dependent |
50 |
5 |
|
Astmul bronşic |
47 |
24 |
|
Luxaţia cong. de şold |
40 |
3 |
|
Despicătura labio-palatină |
38 |
8 |
|
Artrita reumatoidă |
34 |
7 |
|
Piciorul strâmb |
33 |
3 |
|
Hipertensiunea arterială |
25 |
7 |
|
Stenoza pilorică |
15 |
2 |
|
Cancerul cu aceeaşi localizare |
7 |
3 |
|
Spina bifida |
6 |
3 |
Heritabilitatea (simbolizată prin h2) este o noţiune cantitativă ce măsoară amploarea cu care diferitele alele ale unor loci sunt responsabile de variabilitatea (varianţa) unui caracter cantitativ dat, în populaţie sau printre rudele cu un anumit grad de înrudire: părinţi-copii, fratrie, gemeni MZ şi DZ. Ulterior conceptul de heritabilitate a fost extins şi la caractere calitative pentru a exprima cota de participare a eredităţii.
Heritabilitatea este estimată prin gradul de asemănare dintre rude exprimat în forma unui coeficient de corelaţie. O soluţie alternativă de calcul se bazează pe calcularea ratelor de concordanţă la gemenii MZ şi DZ; formula de calcul este:
Concordanţa la MZ – concordanţa la DZ
h2 = –––––––––––––––––
Concordanţa la DZ
Valoarea h2 variază între 0 – dacă genele nu contribuie la variaţia fenotipică totală a caracterului – şi 1, dacă genele sunt responsabile exclusiv de determinismul acelui caracter. Deci, cu cât heritabilitatea este mai mare, cu atât mai mare este contribuţia factorilor genetici la realizarea caracterului respectiv (tabel 5.16). De precizat că acest concept teoretic nu dă informaţii privind numărul de gene implicate sau mecanismul lor de acţiune.
Tabel 5.16 Estimarea heritabilităţii în
diferite boli
|
Boală |
Frecvenţă % |
Heritabilitate |
|
Schizofrenie Astm bronşic Despicături labiale±palatine Stenoză pilorică Spondilită anchilozantă Picior strâmb Boală coronariană Hipertensiune esenţială Luxaţie congenitală de şold Anencefalie şi spina bifida Ulcer peptic Boala congenitală de cord |
1,0 4,0 0.1 0,3 0,2 0,1 3,0 5,0 0,1 0,3 4,0 0,5 |
85 80 76 75 70 68 65 62 60 60 37 35 |
Limitele studiilor gemenilor.
Studiul gemenilor este o metodă ideală pentru evaluarea rolului eredităţii şi mediului în producerea unor boli dar a adus puţine contribuţii majore în genetica umană. Există nu numai limite numerice (puţine cupluri disponibile) ci şi ştiinţifice. Astfel, gemenii MZ nu au exact aceeaşi expresie a genelor cu toate că ei au genotipul identic: de exemplu, inactivarea întâmplătoare a cromosomilor X după clivarea unui zigot în doi gemeni monozigoţi feminini produce diferenţe semnificative în expresia alelelor genelor de pe cromosomul X, în diferite ţesuturi. Apoi, expunerea la factorii de mediu nu este întotdeauna identică pentru gemeni, mai ales la vârsta de adult, odată cu părăsirea căminului copilăriei. Mai mult, chiar şi intrauterin, mediul nu este absolut identic. Gemenii MZ, mono- sau di-placentari pot avea irigare sangvină placentară diferită, dezvoltare intrauterină diferită şi deci greutate la naştere diferită.
c). Studiile de adopţie.
Studiul copiilor adoptaţi este probabil cea mai bună metodă pentru estimarea contribuţiei factorilor genetici în determinismul bolilor multifactoriale. Studiul constă în compararea ratei unor îmbolnăviri la copiii adoptaţi ai unor părinţi afectaţi cu rata de îmbolnăvire a unor copii adoptaţi ai unor părinţi ne afectaţi. De exemplu, copiii adoptaţi ai unor părinţi schizofrenici, fac şi ei boala în proporţie de 8-10%, în timp ce copiii adoptaţi ai unor părinţi ne afectaţi, dezvoltă boala numai în proporţie de 1%. Dificultăţile studiilor de adopţie sunt reprezentate de lipsa frecventă a informaţiilor despre familia biologică, combinată cu plasamentul selectiv al copiilor adoptaţi.
De subliniat că atât studiul gemenilor cât şi studiul adopţiilor sunt metode limitate care nu pot prin ele însele aduce argumente decisive în ce priveşte rolul genelor în determinismul trăsăturile multifactoriale şi nici nu pot identifica genele specifice ale bolii. Aceste studii servesc numai ca indicii preliminare privind rolul componentelor genetice în producerea bolii.
4.2. TEORIILE CARE EXPLICĂ DETERMINISMUL GENETIC AL CARACTERELOR MULTIFACTORIALE
a). Ereditatea poligenică a caracterelor cantitative cu distribuţie continuă (normală sau gaussiană).
Orice caracter fiziologic care poate fi măsurat (talia, greutatea, perimetrul cranian, tensiunea arterială, colesterolul seric, inteligenţa) este un caracter cantitativ. Ereditatea caracterelor cantitative este distinctă de ereditatea monogenică a caracterelor calitative dar poate fi descrisă în termeni mendelieni dacă se acceptă teoria lui Fisher (1918) a eredităţii poligenice. Potrivit acestei teorii:
un caracter cantitativ este determinat de acţiunea simultană a mai multor gene nealele (loci);
genele au efecte cantitative, mici şi aditive/cumulative (spre deosebire de cele monogenice a căror efect este important şi calitativ);
genele acţionează independent unele de altele, între ele nu există relaţii de dominanţă şi recesivitate;
caracterele cantitative poligenice prezintă în populaţie o distribuţie normală, continuă (gaussiană, în formă de clopot) (figura 5.38), spre deosebire de caracterele monogenice a căror distribuţie în populaţie este totdeauna discontinuă;
Curbele de distribuţie normală (gaussiană) se caracterizează prin doi parametri: media (ce determină înalţimea curbei) şi depărtarea faţă de medie (deviaţia standard – DS); circa 68%, 95% şi 99.7% din observaţii se încadrează în medie ± una, două sau trei deviaţii standard.
Expresia fenotipică a caracterelor poligenice poate fi influenţată de factorii de mediu.
Deoarece rudele au gene în comun, funcţiile lor sunt corelate şi mărimea corelaţiei este dependentă de gradul de rudenie.
Efectul aditiv al mai multor gene şi distribuţia normală a caracterului determinat de acestea se pot demonstra cu câteva exemple clasice: pigmentaţia pielii, înălţimea, numărul de creste digitale, tensiunea arterială etc. Înainte de a prezenta unele dintre ele vom preciza faptul că folosirea conceptului de distribuţie normală a unor caractere morfo-fiziologice cantitative este fundamentală pentru medicina clinică, deoarece permite stabilirea unor standarde normale (de vârstă, sex) pentru caracter şi identificarea, prin comparaţie, a unor valori anormale Astfel se conturează un grup de boli multifactoriale definite clinic cantitativ, care sunt de fapt valori extreme (± 2 DS) ale distrinuţie continue a unui caracter normal: hipostatura, obezitatea, unele forme de retard mintal, hipertensiunea arterială esenţială.
Exemplul 1: Pigmentaţia pielii
Caracterul depinde de cantitatea de melanină, un pigment de culoare brună-neagră, depozitat sub formă de granule în celulele tegumentului, în fanere, coroidă. Pigmentaţia pielii variază de la o populaţie la alta; selecţia naturală fixează cea mai favorabilă combinaţie de gene, în funcţie de condiţiile de mediu (ex. populaţia scandinavă din zone cu puţine zile însorite are o foarte redusă cantitate de pigment, în timp ce populaţia de la tropice are, din contră, o mare cantitate de pigment care protejează organismul de radiaţia solară intensă). Caracterul este determinat de un număr de 2-6 gene.
În situaţia cea mai simplă (figura 5.39), presupunem intervenţia a 2 perechi de gene alele: Aa şi Bb, din care genele A şi B determină o cantitate de pigment egală cu 1 iar genele a şi b determină o cantitate de pigment egală cu zero. Negrii, homozigoţi dominanţi (AA ; BB) vor avea o cantitate de pigment egal cu 4, iar albii, homozigoţi recesivi (aa ; bb), o cantitate de pigment egal cu 0. Din căsătoria între un alb şi un negru, rezultă totdeauna mulatri (Aa ; Bb). Din căsătoria între 2 mulatri, pot apărea: negri (N), mulatri închişi (MI), mulatri (M), mulatri deschişi (MD), şi albi (A), în proporţia 1:4:6:4:1 (tabelul 5.17). În ipoteza în care caracterul ar fi determinat de 3 alele independente, proporţia ar fi 1: 6: 15: 20 :15 : 6 : 1, ş.a.m.d.
Tabelul 5.17 Rezultatul combinaţiilor dintre doi mulatri
|
Gameţi |
AB = 2 |
Ab = 1 |
aB = 1 |
ab = 0 |
|
AB = 2 |
AA;BB = 4 N |
AA;Bb = 3 MÎ |
Aa;BB = 3 MÎ |
Aa;Bb = 2 M |
|
Ab = 1 |
AA;Bb = 3 MÎ |
AA;bb = 2 M |
Aa;Bb = 2 M |
Aa;bb = 1 MD |
|
aB = 1 |
Aa;BB = 3 MÎ |
Aa;Bb = 2 M |
aa;BB = 2 M |
aa;Bb = 1 MD |
|
ab = 0 |
Aa;Bb = 2 M |
Aa;bb = 1 MD |
aa;Bb = 1 MD |
aa;bb = 0 A |
Faptul că în prima generaţie apar numai mulatri, ne-ar putea face să credem că pigmentaţia este controlată de o singură pereche de gene. Dacă ar fi aşa, atunci, în generaţia a II-a, conform legilor mendeliene, ar trebui să apară un procent egal de albi şi negri. Dar raportul observat este diferit, singura explicaţie plauzibilă fiind aceea că pigmentaţia este condiţionată de 2 sau mai multe gene, având efect aditiv, numărul lor exact nefiind încă cunoscut. Printre genele candidat se numără cele ce codifică receptorii membranri pentru melanocortină şi proteina melanosomală P, precum şi genele pentru tirozinazele melanogenice TRP1 şi TRP2.
În realizarea culorii pielii intervin însă, în afară de cantitatea de pigment, determinată de un număr mai mare de gene, şi alţi factori ce ţin de individ (sex, vârstă, grosimea tegumentului…), sau de mediu (alimentaţie, lumina solară…). De aceea determinismul acestui tip de caractere este numit, în mod obişnuit, poligenic, multifactorial.
Exemplul 2: Înălţimea populaţiei
Reprezentarea grafică a înălţimii populaţiei este o curbă de tip gaussian, caracteristică distribuţiei continue, normale (figura 5.40). Se poate vedea că faţă de înălţimea medie de 170 cm., valorile sunt repartizate simetric de o parte şi alta a mediei, curba de distribuţie luând aspect de clopot. Care este explicaţia acestei distribuţii?
Presupunem (ne-realist) că înălţimea ar fi determinată de o singură genă cu 2 alele A şi a. Alela A tinde să producă indivizi cu talie înaltă, alela a indivizi cu talie joasă. Dacă nu există o dominanţă la acest locus, cele 3 genotipuri posibile (AA, Aa, aa) vor produce 3 fenotipuri: înalt, mediu şi scund. Presupunând că frecvenţa genelor A şi a în populaţie este 0,5 pentru fiecare, distribuţia, grafic reprezentată, va fi cea din figura 5.41.a.
Presupunem acum, mai realist, că talia este determinată de doi loci, cel de al doilea de asemenea cu două alele: B pentru talia înaltă şi b pentru talia scundă. Ele influenţează talia exact ca şi alelele A şi a. Acum sunt 9 posibilităţi genotipice şi 5 posibilităţi fenotipice după cum individul va avea 0, 1, 2, 3 sau 4 alele pentru talia înaltă. Distribuţia în populaţie încă nu este una normală dar este mult mai apropiată de o distribuţie normală decât în cazul anterior cu o singură genă. (figura 5.41.b)
Şi, în sfârşit, dacă extindem exemplul la situaţia în care mai multe gene şi factori de mediu influenţează înălţimea, fiecare având un efect redus, rezultă mai multe fenotipuri posibile, fiecare puţin diferit de celălalt, iar distribuţia în populaţie dezvoltă o curbă în formă de clopot. (figura 5.41c)
Din exemplele prezentate se poate concluziona că un fenotip care are o distribuţie normală în populaţia generală poate fi produs de ereditatea poligenică, implicând acţiunea mai multor gene, cu efecte aditive. Trebuie subliniat faptul că genele individuale care stau la baza unor caractere cantitative poligenice, multifactoriale, se transmit conform principiilor mendeliene ale segregării şi asortării independente. Singura deosebire este aceea că ele acţionează împreună pentru a determina caracterul respectiv.
Pentru efectul aditiv al mai multor gene pledează şi studiul corelaţiilor familiale pentru caractere cantitative. Corelaţia este o măsură statistică a gradului de asociere al unor fenomene variabile sau, mai simplu, o măsură a gradului de asemănare sau de relaţie între doi parametri. De exemplu, pornind de la faptul că rudele de gradul I au 50% din gene în comun se poate presupune că un caracter este poligenic (talie, inteligenţă etc) atunci corelaţia dintre rude le de gradul I, ca de exemplu părinţi-copii, frate-frate, ar trebui să fie 0,5. Această premisă teoretică a fost validată de studiile care au arătat o corelaţie frate-frate apropiată de 0,5 pentru înălţime, QI, număr de creste digitale. Totuşi, diferite studii ale unor caractere cantitative au evidenţiat „o regresie faţă de medie”. De exemplu, părinţi care sunt înalţi sau inteligenţi au copii a căror înălţime sau inteligenţă medie este mai mică decât media valorilor parentale; dacă aceste caractere ar fi pur poligenice atunci datele obţinute la descendenţi ar trebui să se plaseze în jurul mediei valorilor parentale. Or, în realitate, caracterele umane cantitative pot fi influenţate de mediu şi este de asemenea posibil ca unele gene implicate să nu aibe efecte aditive şi să acţioneze dominant. Toate acestea sugerează existenţa altor modele de explicaţie a eredităţii multifactoriale.
b). Ereditatea poligenică cu prag a caracterelor discontinue.
În genetica medicală multe malformaţii congenitale şi boli comune ale adultului (tabelul 5.12) au o distribuţie familială şi nu prezintă un model de transmitere monogenică. De aceea, firesc, s-a presupus că ele sunt poligenice, multifactoriale, dar – spre deosebire de caracterele cantitative care au o distribuţie continuă în populaţie – ele prezintă o distribuţie discontinuă (fiind prezente sau absente la o persoană dată, întocmai ca şi caracterele monogenice). Pentru a explica producerea acestor caractere multifactoriale discontinue, Falconer (1971) a extins teoria poligenică, postulând existenţa unei susceptibilităţi variabile la boală, cu o distribuţie continuă în populaţie care prezintă însă un prag care trebuie trecut, depăşit, pentru ca boala să se manifeste (figura 5.42). Sub prag, individul este aparent normal; peste prag el poate fi afectat.
Modelul poligenic cu prag al susceptibilităţii la boală presupune că bolnavii au moştenit o combinaţie „nefericită” de gene de risc. Deoarece rudele lor posedă un număr de gene în comun este logic să presupunem că au şi ele o susceptibilitate crescută, un risc mai mare de boală, comparativ cu populaţia generală; de aceea bolile respective au un caracter familial. Astfel, urmărind proporţia rudelor afectate în familii cu despicătură labio-palatină (tabelul 5.17) se poate observa că:
proporţia gemenilor monozigoţi afectaţi este de circa 40%, comparativ cu proporţia de 4% pentru gemenii dizigoţi;
proporţia pentru indivizii cu copii afectaţi este identică cu cea pentru fraţi afectaţi;
există o scădere netă a proporţiei rudelor afectate, în funcţie de gradul de rudenie: o scădere de 10 ori de la gemeni monozigoţi la gemeni dizigoţi, o scădere de 7 ori de la rude de gradul I la rude de gradul II, o scădere de 2,5 ori de la rudele de gradul II la rudele de gradul III. Deci, frecvenţa bolii multifactoriale în familie scade cu reducerea gradului de înrudire, ajungând a fi identică cu cea din populaţia generală
Tabelul 5.18 – Proporţia rudelor afectate în despicătura labio-palatină (Chen, 1988)
|
Grad de rudenie |
Afectaţi |
Proporţia de afectaţi |
Factorul de risc faţă de populaţia generală |
|
Gemeni |
MZ DZ |
~ 40% 4% |
|
|
Gradul I |
Fraţi Copii |
3,2 – 4,9% 3,0 – 4,3% |
40 x |
|
Gradul II |
Mătuşi şi unchi Nepoţi şi nepoate |
0,6 – 0,8% 0,7 – 0,8% |
7 x |
|
Gradul III |
Veri primari |
0,3% |
3 x |
|
Neînrudit |
Populaţia generală |
0,1% |
1 x |
Aceste date ne permit să prezentăm modelul susceptibilităţii cu prag (figura 5.42). Conform acestui model, toţi factorii care influenţează dezvoltarea unor afecţiuni multifactoriale, fie ei genetici sau de mediu, pot fi consideraţi ca o singură entitate, cunoscută ca susceptibilitate. Susceptibilitatea tuturor indivizilor într-o populaţie formează o variabilă continuă care are o distribuţie normală atât în populaţia generală cât şi la rudele individului afectat. Curba de distribuţie prezintă însă un prag dincolo de care indivizii au mai multe gene de risc şi deci o predispoziţie mai mare de boală. Factorii de mediu vor determina uneori când şi cât de afectate vor fi persoanele cu predispoziţie genetică. Boala apare atunci când o persoană cu un număr important de gene de risc (depăşeşte deci pragul de risc), se expune unor condiţii defavorabile de mediu, care transformă predispoziţia genetică în boală.
Atingerea şi depăşirea pragului de risc depinde de numărul de gene de risc pe care o persoană le moşteneşte de la ambii părinţi. În consecinţă, în bolile multifactoriale cu predispoziţie genetică se observă următoarele caracteristici:
boala are frecvent un caracter familial; cu cât afecţiunea este mai rară în populaţia generală, cu atât boala este mai frecventă printre rude (de exemplu, despicăturile labiale – cu o frecvenţă în populaţia generală de 2/1000 au o frecvenţă de 3% printre rudele de gradul I; despicăturile palatine –1/1000 – au o frecvenţă de 5% printre rudele de gradul I)
indivizii afectaţi se găsesc dincolo de pragul distribuţiei în populaţie a susceptibilităţii determinate genetic, dar poziţia lor exactă nu poate fi definită; de aceea, pentru cele mai multe boli multifactoriale s-a stabilit un risc empiric, bazat pe analiza unui număr mare de familii în care apare un bolnav (în bolile mendeliene riscul este determinat de modelul specific de transmitere ereditară, AD sau AR sau LX).
părinţii bolnavului pot fi sănătoşi sau unul dintre ei poate fi bolnav;
un bolnav are un risc mai mare de a avea copii bolnavi comparativ cu persoanele din populaţia generală;
rudele de gradul I ale bolnavului (fraţi, copii) au un risc mai mare de boală, comparativ cu populaţia generală; riscul scade însă odată cu scăderea gradului de rudenie (figura 5.43)
riscul de îmbolnăvire diferă de la o familie la alta, funcţie de numărul de gene de risc (spre deosebire de transmiterea monogenică în care riscul este constant); în plus, el poate fi diferit în populaţii diferite datorită faptului că frecvenţele genelor implicate ca şi a factorilor de mediu sunt diferite. De exemplu, riscul empiric pentru defectele de tub neural în SUA este de 2-3%, în timp ce în populaţia britanică este de 5%.
Aceste caracteristici sunt utile pentru stabilirea naturii multifactoriale ale unor boli în care factori genetici multipli determină o predispoziţie la boală iar mediul este cel care transformă vulnerabilitatea în boală. În practică, diagnosticul de ereditate multifactorială este un diagnostic de excludere. Trăsăturile clinice ale unei boli multifactoriale nu se deosebesc de obicei de trăsături similare de altă cauză. De exemplu, DLP, singură sau asociată cu alte anomalii este, obişnuit, multifactorială. Dar sunt peste 200 de afecţiuni în care DLP este doar o manifestare în cadrul unui sindrom plurimalformativ specific. Multe din aceste condiţii sunt consecinţa unor anomalii cromosomiale, altele ale unor boli monogenice, iar altele ale unor factori de mediu teratogeni. Uneori este dificil de a distinge o boală poligenică de una monogenică cu penetranţă redusă sau expresivitate variabilă.
Deoarece factorii de risc sunt diferiţi pentru diferite boli multifactoriale, riscul empiric de recurenţă este şi el specific fiecărei boli în parte, fiind dependent şi de frecvenţa bolii în populaţie. Riscul de recurenţă pentru rudele de gradul I, adică fraţi-surori sau copii, este aproximativ egal cu rădăcina pătrată a incidenţei generale în populaţie. Astfel, pentru malformaţiile congenitale izolate, care au o incidenţă în populaţie de circa 1/1000, dacă doi părinţi sănătoşi, fără alte antecedente familiale, au un copil afectat, riscul de recurenţă la sarcinile următoare este de 1 la 32 sau 3%; acelaşi risc îl au şi descendenţii unui bolnav. Pentru bolile multifactoriale ale adultului, care au în populaţie o prevalenţă de circa 1/100, riscul de recurenţă în fratrie şi la copiii unui individ afectat este de 10%.
Riscul la descendenţi creşte (peste 5% în malformaţiile congenitale izolate) dacă:
în familie sunt mai multe persoane afectate (în bolile monogenice riscul este constant); de exemplu, în cazul despicăturii labiale ± palatine (DLP) riscul de recurenţă după naşterea unui copil afectat este 4 % iar dacă sunt doi copii afectaţi riscul creşte peste10 %); prezenţa unui al doilea bolnav în familie nu modifică per se riscul genetic ci indică faptul că familia are o predispoziţie gentică mai mare şi deci un risc de recurenţă mai mare
bolnavul prezintă o formă mai gravă de boală[46]; de exemplu, riscul de recurenţă în DLP unilaterală este de 2,5%, în cea bilaterală riscul este de 6%; Cu cât defectul este mai sever cu atât susceptibilitatea genetică este mai mare;
dacă unul din sexe este mai frecvent afectat (ex. sexul masculin pentru stenoza congenitală de pilor sau sexul feminin pentru luxaţia de şold), riscul de recurenţă este mai mare după naşterea unui copil care aparţine sexului mai rar afectat, deoarece individul afectat, de sex mai puţin susceptibil, este obişnuit într-o poziţie extremă a distribuţiei de susceptibilitate. Astfel, în stenoza de pilor cu sex-ratio de 5M:1F, dacă tatăl a fost afectat, recurenţa este de 5,5% la băieţi şi de 2,4% la fete; dacă mama a fost afectată, riscul este de 19,4% pentru băieţi şi de 7,3% pentru fete.
au loc căsătorii consangvine şi persoanele înrudite au un număr mai mare de gene în comun.
c). Ereditatea oligogenică sau modelul mixt
Chiar dacă modelul susceptibilităţii cu prag descris anterior este satisfăcător prin criteriile pe care le oferă, pentru unele boli multifactoriale pot fi şi alte explicaţii. Este posibil ca diferite gene implicate în boală să joace roluri de importanţă inegală. În unele cazuri, un caracter poate fi determinat de acţiunea combinată a unei /unor gene de susceptibilitate cu efect major şi a unui “fond” multifactorial în care genele adiţionale şi factorii de mediu au fiecare un mic efect adiţional.
Dacă luăm din nou ca exemplu talia, putem presupune că variaţia în înălţime este determinată de un singur locus (sau genă majoră) şi o componentă multifactorială. Indivizii cu genotipul AA vor tinde spre o talie înaltă, cei cu genotip aa spre o talie mică iar cei cu genotip Aa spre o talie intermediară. Astfel, cei cu genotip aa vor avea variaţii ale taliei între 130 cm şi 170 cm., cei cu genotip Aa vor avea variaţii ale taliei între 150 cm şi 190 cm. iar cei cu genotip AA vor avea variaţii ale taliei între 170 cm şi 210 cm. (figura 5.44).
O explicaţie utilă e oferită de dr. Michael Brown de la Universitatea din Texas care face o analogie cu ceea ce el numeşte “sindromul cucuiului”. Dacă s-ar reduce imaginar tocul uşilor de la 200 cm. la 170 cm. am avea de a face cu aşa-zisul “sindrom al cucuiului”. Boala ar surveni rar în familiile cu înălţime medie dar ar atinge proporţii epidemice la familiile înalte. Tocul uşii converteşte distribuţia continuă a înălţimii în aceste familii în două fenotipuri: cu cucui, cei peste 170 cm. (zona neagră din figura 5.38) şi fără cucui, cei sub 170 cm. (zona haşurată din figura 5.39). Deci chiar dacă nivelul prag rămâne acelaşi, numărul indivizilor cu cucui este proporţional mai mare decât cel aşteptat doar prin determinismul monogenic şi asta datorită faptului că lor li se adaugă cei cu predispoziţie genetică prin cumulul mai multor factori (genetici sau de mediu).
Din cauza influenţei „fondului” multifactorial, există o substanţială suprapunere ale celor trei genotipuri majore. Distribuţia globală a înălţimii, sub formă de clopot, este determinată de superpoziţia celor trei distribuţii ale fiecărui genotip.
Acest model în care factorii genetici sunt reprezentaţi de o componentă monogenică (o genă majoră, cu mai multe alele), o componentă poligenică şi factori de mediu, se numeşte model mixt. În acest caz distribuţia susceptibilităţii la boală este rezultanta mai multor distribuţii gaussiene corespunzând diferitelor genotipuri posibile (3 dacă sunt 2 alele) ale locusului major.
Teoria oligogenică ar reprezenta de fapt o piesă a spectrului complex al determinismului caracterelor umane, ilustrat în figura 5.45, care se plasează în diferite poziţii faţă de cei trei poli: monogenic, poligenic şi ecologic. Această teorie deschide o perspectivă optimistă pentru identificarea genelor de predispoziţie la boală
4.3. IDENTIFICAREA GENELOR IMPLICATE ÎN BOLILE MULTIFACTORIALE
Bolile multifactoriale – fie că este vorba de malformaţii congenitale sau de boli comune ale adultului – sunt frecvente şi au o contribuţie majoră la morbiditate şi mortalitate. Este deci firesc să se facă eforturi importante pentru a identifica genele care contribuie la etiologia lor, determinând susceptibilitatea la boală. In cazul caracterelor mendeliene, datorită corelaţiei puternice care există între genotip şi fenotip, studiile de înlănţuire permit de regulă cartografierea caracterului în interiorul unui interval minim de ordinul a 1cM. Ulterior sunt identificate variaţiile de secvenţă prezente la indivizii afectaţi în regiunile codante ale genelor candidat şi aceasta permite dovedirea corespunzătoare a identităţii genei implicate. Acest model de lucru nu se aplică însă în cazul caracterelor multifactoriale, în general pentru identificarea genelor implicate fiind necesare patru etape majore de lucru:
(1). Studiile de înlănţuire şi asociere.
Acestea au ca scop stabilirea unei asocieri semnificative a unei anumite regiuni genomice cu caracterul cantitativ respectiv. Cel mai adesea însă, chiar şi în cazul unor studii pe colecţii mari de familii, intervalul minim nu poate fi restrâns sub dimensiuni de 10-30cM, ceea ce corespunde la om unui fragment de 10-30Mb sau circa 100-300 gene.
Studiile de asociaţie compară incidenţa unui anumit polimorfism la bolnavi cu incidenţa ei într-un grup control; dacă o alelă specifică a locusului polimorfic are o prezenţă semnificativ mai mare la indivizii cu boală comparativ cu cei neafectaţi atunci se poate presupune că alela respectivă este implicată în patogenia bolii. Astfel s-a putut stabili că alela epsilon 4 a genei apolipotpoteinei E poate fi un factor important de risc pentru boala Alzheimer.
Studiile de înlănţuire analizează cotransmisia în familii a unui locus marker şi a locusului boală. Aceasta ar aduce dovezi asupra implicării locusului respectiv (sau a altuia situat foarte aproape de el) în procesul de boală. Cel mai frecvent se foloseşte analiza de înlănţuire a unei perechi de membri (frati) ai unei familii care sunt concordanţi pentru boală, deci au foarte probabil în comun anumite alele predispozante; se recurge la scanarea genomului (cu ajutorul a sute de markeri ADN polimorfici) pentru a găsi regiuni prezente la ambii membri mai frecvent decât s-ar aştepta; aceasta permite prezumţia ca în regiunea respectivă se află un locus implicat în boală.
(2). Cartografierea de fineţe.
Această etapă are ca scop reducerea la maximum posibil a dimensiunii intervalului critic şi se realizează prin studii familiale de dezechilibru a înlănţuirii (linkage disequilibrium – LD) sau prin studii caz-control. Asemenea studii permit reducerea intervalului critic la dimensiuni de ordinul a 1 cM.
(3). Analizele secvenţelor.
Analiza secvenţei ADN în intervalul minim este necesară pentru identificarea variantelor nucleotidice prezente. În ciuda eforturilor intervalul minim poate conţine frecvent câteva gene, fiecare cu numeroase variante nucleotidice, unele la nivelul regiunilor codante, iar altele în regiunile care le flanchează. Se considera că adeseori caracterele multifactoriale pot fi determinate mai degrabă de variante ale unor secvenţe reglatorii decât de variante în secvenţele codante. Dacă în cazul regiunilor codante efectele pot fi uşor prevăzute prin simpla corelaţie cu codul genetic, consecinţele unor modificări în regiunile necodante sunt mult mai dificil de interpretat.
(4). Teste funcţionale ale variantelor nucleotidice candidate.
Aceasta etapă este complet diferită faţă de analizele utilizate pentru identificarea caracterelor mendeliene şi constă în dovedirea pe cale experimentală a efectului funcţional al variantelor candidat identificate. Aceasta presupune utilizarea unor modele animale sau culturi de celule umane şi se bazează pe tehnologia knock-in sau prin analiza efectului pe care îl are introducerea prin inginerie genetica a variantei nucleotidice testate. De multe ori este insa dificilă extrapolarea rezultatelor între specii diferite.
In ciuda dificultatilor menţionate au fost identificate câteva variante genice implicate în etiologia unor caractere multifactoriale la om. Mai jos sunt descrise câteva dintre cele mai sugestive exemple.
Astfel, înlănţuirea genetică a diabetului zaharat tip 1 cu genele complexului major de histocompatibilitate HLA-DR si HLA-DQ a fost stabilită de peste 20 de ani. Există mai multe categorii de dovezi care suportă ferm rolul alelei DQ-beta în susceptibilitatea la DZ tip 1: conservarea între specii a aminoacidului din poziţia 57; asocierea semnificativă între DQ-beta şi DZ tip 1 în diverse populaţii; faptul că aminoacidul 57 este structura cheie a moleculei DQ-beta.
In diabetul zaharat tip 2 un screening la nivelul întregului genom a permis identificarea unei regiuni de pe cromosomul 2 puternic înlanţuită cu boala. Regiunea a putut fi redusa din fericire la circa 1,7 Mb. În interiorul acestei regiuni au fost identificate multiple polimorfisme mononucleotidice (SNP), iar dintre acestea, haplotipul unei grupări de trei SNP de la nivelul genei pentru calpacaina-10 (CAPN10) s-a dovedit a fi puternic asociat cu DZ tip 2 în mai multe populaţii.
Alela E4 a apolipoproteinei E (APOE4) este asociată într-o manieră doză-dependenta cu susceptibilitatea la boala Alzheimer. Aceasta asociere, împreuna cu localizarea proteinei APOE4 la nivelul leziunilor cerebrale şi cu rolul proteinei ApoE (de la şoarece) în formarea depozitelor de amiloid susţin cu putere rolul APOE4 în susceptibilitatea la boala Alzheimer.
Gena CARD15 (care codifică proteina 15 cu domeniu de recrutare a caspazei) localizată la nivelul cromosomului 16 a fost identificată ca genă de susceptibilitate la boala Crohn. In interiorul acestei gene au fost identificate câteva variante frameshift şi cu sens gresit care sunt puternic asociate cu boala, deşi confirmarea efectului lor prin studii knock-in la soareci încă lipseşte.
Alte gene implicate în etiologia unor caractere multifactoriale pentru care există dovezi puternice sunt ADAM33 (cromosomul 20, codifică o proteina din familia dezintegrinelor si metaloproteazelor) în astmul bronşic sau ACE1 (cromosomul 17, codifica enzima de conversie a angiotensinei 1) in susceptibilitatea la hipertensiunea arteriala esentiala.
INTERNET
Dicţionar de biologie celulară: http://www.mlab.gla.ac.uk/~julian/Dict.html
Genetică medicală – University of Utah School of Medicine: http://medgen.genetics.utah.edu
Genetică medicală – http://www.hslib.washington.edu-helix
Gene amprentate; http://www.mgu.har.mrc.ac.uk/imprinting
Principii de genetică -Indiana U niversity Biotechnology: http://biotech.chem.indiana.edu
National Human Genome Research Institut: http://www.nhgri. nih.gov.
Nomenclatura genelor: http://www.gene.ucl.ac.uk/cgi-bin/nomenclature/searchgenes.pl
NORD: asociaţia naţională pentru boli rare: http://www.NORD-rdb.com/~orphan
OMIM: http://www3.ncbi.nlm.nih.gov/omim
Bibliografie specifică selectivă
Badano, J.L, Katsanis, N. – Beyond Mendel: an evolving view of human genetic disease transmission – Nat. Rev. Genet. 2002;3:779-789.
Barlow D.P. – Gametic imprinting in mammals – Science 1995;270:1610-1613
Barsh GS. – The genetics of pigmentation: from fancy genes to complex traits – Trends Genet., 1996;12:299 -305
Bernards A., Gusella JF. – The importance of genetic moyaicism in human disease – N.Engl.J.Med.,1994; 331:1447-1449
Dynalcht BD – Regulation of transcription by proteins that control the cell cycle – Nature,1997;389:149-152
Gao CY, Zelenka PD. – Cyclins, cyclin-dependent kinases and differentiation – BioEssay, 1997;19:307-315.
Glazier, A.M., Nadeau, J.H., Altman T.J. – Finding genes that underlie complex traits – Science, 2002;298:2345-2349.
Ghosh S., Collins FS. – The geneticist’s approach to complex disease – Ann.Rev.Med. 1996;47:333-353
Hall JG. – Twins and twinning – Am.J.Med.Genet., 1996;61:202-204.
Hall JG. – Genomic imprinting: nature and clinical relevance – Ann.Rev.Med., 1997; 48:35-44.
Hawkes CH. – Twin studies in medicine-what do they tell us? – QJ Med. 1997;90:311-321.
Hickey RJ; Malkas LH – Mammalian cell DNA replication – Crit. Rev.Eukar. Gene Expres. 1997;7:125-157
Lander E., Kruglyak L. – Genetic dissection of complex traits: Guidelines for interpreting and reporting linkage results – Nat.Genet., 1995;11:241-247
Latham KE. – X chromosome imprinting and inactivation in the early mammalian embryo – Trends Genet. 1996;12:134-138
Lee JT, Jaenisch R – The (epi)genetic control of mammalian X-chromosome inactivation – Curr.Opin.Genet.Dev., 1997;7:274-280.
Matin, A., Nadeau, J.H. – Sensitized polygenic trait analysis – Trends Genet. 2001;17: 727-731.
Nadeau J.H. (2001) – Modifier genes in mice and humans – Nat. Rev. Genetics, 2: 165-174.
Reddy PS, Houssman DE. – The complex pathology of trinucleotid repeats – Curr.Opin.Genet.Dev., 1997;9:365-372.
Roeder G.S. – Meiotic chromosomes: it takes two to tango – Genes & Development, 1997, 11:2600-2621
Romeo G., McKusick V.A. – Phenotypic diversity, alelic series and modifier genes – Nature Genet 1994;7:451-453
Risch N, Merikangas K. – The future of genetic studies of complex human diseases – Science 1996;273:1526-1517
Tjian R. – Molecular machines that control genes – Sci.Amer. 1995; 272:54-61.
Zlotogora J. –Germ line mosaicism – Hum.Genet., 1998;102:381-386.
Wilkie AOM. – The molecular basis of dominance – J.Med.Genet., 1994;31:89-98
Warburton D. – Human female meiosis: new insight into an error-prone process – Am.J.Huma.Genet. 1997; 61:1-4
[1] Demonstrarea experimentală a replicării semiconservativea fost realizată, cu eleganţă şi precizie, prin încorporarea unor nucleotide marcate radioactiv (Meselson şi Sthal,1957; Taylor, 1957) sau a bromodeoxiuridinei în moleculele de ADN care se sintetizează (Zaharov, 1972; Latt, 1973)
[2] RNaza H1 degradează specific hibridul format de amorsa ARN şi ADN (numele H provine de la Hybrid)
[3] Piesele Okazaki se vor sintetiza în direcţia 5’® 3’, care va fi în direcţia opusă furcii de replicare.
[4] Repetiţiile multiple ale dinucleotidului AT sunt asociate frecvent cu regiunile de ataşare la “axul “– intern al cromosomului – (“scaffold”) şi codifică elemente specifice necesare destabilizării structurii helicale a ADN; de aceea sunt numite şi “DNA unwiding elemnts” (DUEs)
[5] Ciclinele D, E şi A împreună cu Cdk4 alcătuiesc SPF (S-phase promoting factor) care controlează faza S
[6] După Greider, 1999, se formează chiar o structură triplu catenară, asemănătoare cu bucla D de la ADNmitocondrial
[7] la om, celulele somatice pot produce prin diviziune maximum 80 de generaţii celulare (ipoteza Hayflick); (excepţie fac însã celulele stem pluripotente)
[8]Datoritã numãrului mare de diviziuni ce au loc în spermatogenezã, absenţa telomerazei ar duce la extincţia rapidã a acestei preţioase populaţii celulare .
[9]De aceea dispariţia postnatalã a activitãţii telomerazei ar putea fi consideratã un "mecanism de prevenire" a proliferãrii excesive şi anormale a celulelor somatice.
[10]Unele cancere care necesitã puţine mutaţii pentru a deveni maligne şi deci nu implicã decât un numãr mic de diviziuni celulare ar putea fi telomeraz negative
[11]Oligonucleotide modificate de tipul "peptide nucleic acids (PNAs) (7) în care deoxiriboza a fost înlocuitã de N-glicinã
[12] Ciclul celular meiotic are o serie de particularităţi (vezi capitolul 5.C.1)
[13] Poziţia punctului de oprire G1 este incertă, fie aproape de faza S, fie la mijlocul fazei G1
[14] Moleculele CKI (de le Cyclin-dependentKinase Inhibitors) sunt de două feluri: KIP (de la kinase inhibitory proteins) care acţionează pe multiple complexe Cdk-ciclină şi INK (de la Inhibitors of Cdk) care acţionează specific pe complexul Cdk-ciclina D
[15] Proteina Rb este codificată de o genă supresoare de tumori situată pe 13q14; mutaţiile genei Rb pot produce anumite cancere, inclusiv retinoblastoame.
[16] Gena tp53 este o genă supresoare de tumori; mutaţiile ei se regăsesc ăn peste 60% din cancere
[17] PCNA stimulează ADN polimeraza δ
[18] Acest complex produce fosforilarea proteinelor implicate în despiralizarea ADN precum şi a proteinei RPA care se fixează în punctele de origine ale replicării. După declanşarea replicării complexul RPA-origine este blocat de către Cdk-ciclinaB şi replicarea nu se poate repeta (se face o singură dată) pâna ce celula nu trece prin mitoză, care degradează complexul Cdk-ciclina B
[19] Cdc2 este o Cdk specifică, numită aşa de la cell division cycle mutant 2
[20] Să reamintim că metafaza este stadiul optim pentru studiul cromosomilor, deoarece aceştia sunt contractaţi, bine individualizaţi morfologic şi dispuşi într-un singur plan.
[21] Funcţia microtubulilor poate fi demonstrată prin tratament cu colchicină, care inhibă formarea lor şi împiedică aranjarea cromosomilor în plan ecuatorial, precum şi migrarea lor anafazică.
[22] In acest context, termenul de “reproducere” este impropriu pentru că individul ce rezultă după fecundare nu este o reproducere a nici unuia din părinţi.
[23] Există două diferenţe majore între meioză şi mitoză: prin meioză rezultă celule haploide, care genetic sunt diferite
[24] Alinierea / împerecherea cromosomilor omologi “margine” la “margine” la nivelul sinapselor este favorizată de secvenţele repetitive multiple şi identice ca tip şi localizare din cei doi cromosomi omologi.
[25] Dispoziţia cromomerelor coincide cu cea a benzilor G (vezi capitolul Cromosomii umani)
[26] Mecanismul molecular al recombinării la om nu este prea clar dar se presupune ca intervine un sistem genic similar cu genele „SOS” descrise la procariote, care prin enzimele codificate taie, schimbă şi resudează fragmentele de cromosomi.
[27] MITE (de la mariner insect transposon-like element) are o structură similară cu transposonul mariner de la insecte. MITE nu este însă un transposon activ; în genomul uman au mai fost identificate si alte secvenţe tip MITE dar originea lor este misterioasă.
[28] În această situaţie, când ne referim la cromosomi avem în vedere centromerele deoarece o parte din materialul lor s-a schimbat prin CO, deci fiecare cromosom are şi segmente materne şi paterne.
[29] S-a discutat şi reducerea competenţei „imunologice” dintre mamă şi făt odată cu creştere vârstei materne, fapt ca ar permite supravieţuira embrionilor trisomici; lipsesc probe pentru această ipoteză.
[30] Himera se deosebeşte de mozaic deoarece populaţiile (clonele) celulare diferite au origini diferite; în mozaicuri, clonele celulare anormale au origine comună cu cele normale, provin din acelaşi zigot.
[31] Se mai numeşte legea disjuncţiei caracterelor parentale sau legea purităţii gameţilor.
[32] Negustătorul este asimilat tipului parental şi atunci încrucişarea F2 (sau F1) x P se numeşte retroîncrucişare.
[33] Se mai numeşte şi legea liberei combinaţii a factorilor ereditari sau “legea segregării independente a perechilor de caractere”.
[34] „Pedigree” derivă din expresia franceză „pied de grue” (picior de cocor) care exprimă aspectul ramificat al diagramei
[35] Excepţie fac genele „paseudoautosomale” care au alele funcţionale atât pe X cât şi pe Y
[36] Se recomandă ca alelele ce determină cracatere dominante să fie notate cu majuscule iar alelele ce determină caractere recesive cu litere mici. Noi preferăm simbolurile A sau a, pentru genele mutante şi N sau n pentru cele normale.
[37] Circa 40% din indivizii care fac TVP sunt heterozigoţi pentru alela factorului V Leiden
[38] După adunarea datelor se impune o prelucrare şi o corecţie statistică pentru a compensa pierderea de informaţii determinată de fratriile ce provin din căsătorii între heterozigoţi şi care nu au nici un bolnav; altfel, proporţia de homozigoţi afectaţi va fi mai mare de 25%.
[39] Se realizează aspectul unui caz izolat/sporadic dar ar fi o greşeală să considerăm că boala nu este ereditară numai pentru că nu sunt alte cazuri în familie.
[40] Genele omologe de pe X şi Y pledează pentru originea filogenetică a cromosomilor sexuali dintr-o pereche de autosomi omologi.
[41] Combinaţia XaY + XaXN este posibilă, cu o probabilitate de 1:5.000, şi în cazul hemofiliei dar se pare că hemofilia în stare heterozigotă este letală la femei.
[42] Pacientele cu incontinentia pigmenti tipul 2 prezintă hiperpigmentări cutanate striate, în vârtejuri, absenţa dinţilor, căderea părului şi uneori tulburări neurologice sau microcefalie cu retard mental; supravieţuirea bolnavelor se corelează cu inactivarea preferenţială a XA în marea majoritate a celulelor.
[43] Alela maternă este represată , prin metilare
[44] Utilizarea acestui parametru – prevalenţa în populaţie – este importantă deoarece o boală frecventă în populaţie are şanse mai mari să apară prin coincidenţă la mai mulţi membri din familie
[45] . O diviziune apărută foarte târziu, după a 14-a zi de la concepţie poate da naştere la gemeni uniţi (care apar cu o frecvenţă de 1/50000 de sarcini) cum sunt siamezii…numiţi astfel în memoria celebrilor Chang şi Eng, născuţi în 1811 în Thailanda (Siam) şi uniţi prin abdomenul comun.
